

Integrin-linked kinase functions as a downstream signal of platelet-derived growth factor to regulate actin polymerization and vascular smooth muscle cell migration
- PMID: 20178627
- PMCID: PMC2838830
- DOI: 10.1186/1471-2121-11-16
Integrin-linked kinase functions as a downstream signal of platelet-derived growth factor to regulate actin polymerization and vascular smooth muscle cell migration
Abstract
Background: Vascular smooth muscle cell migration and accumulation in response to growth factors extensively contribute to the development of intimal thickening within the vessel wall. Cumulative evidence has shown that actin cytoskeleton polymerization and rearrangement are critical steps during cellular spreading and migration. Integrin-linked kinase, an intracellular serine/threonine kinase, is a cytoplasmic interactor of integrin beta-1 and beta-3 receptors regulating cell-cell and/or cell-extracellular matrix interaction, cell contraction, extracellular matrix modification, and cell spreading and migration in response to various stimuli. However, the regulatory role of ILK during vascular smooth muscle cell migration and the importance of integrin signaling in occlusive vascular diseases are not yet fully elucidated.
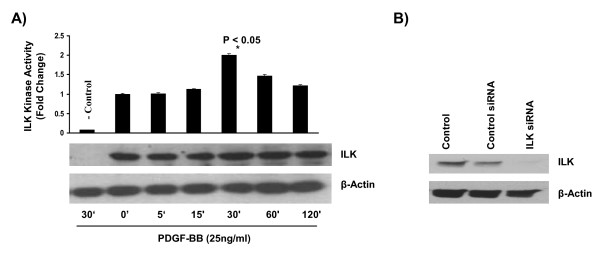
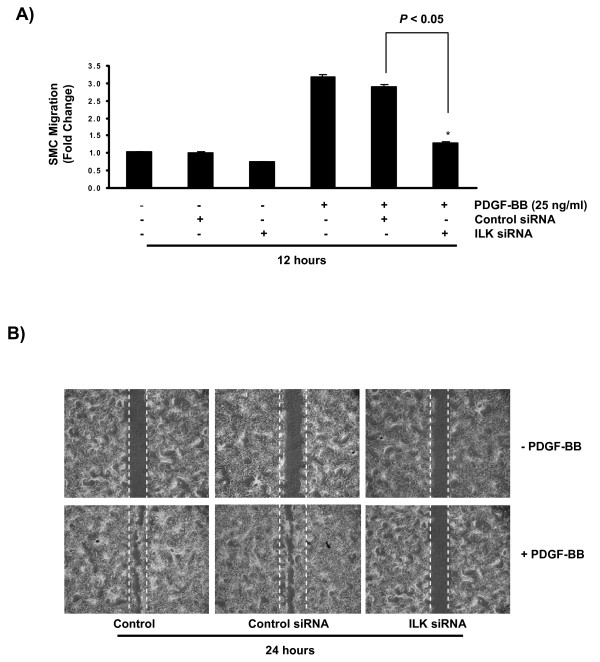
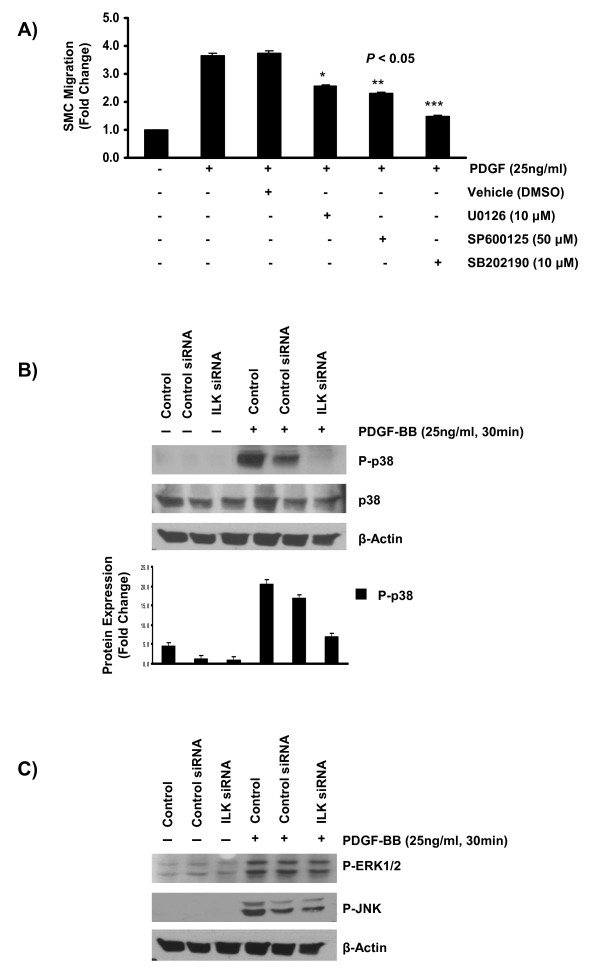
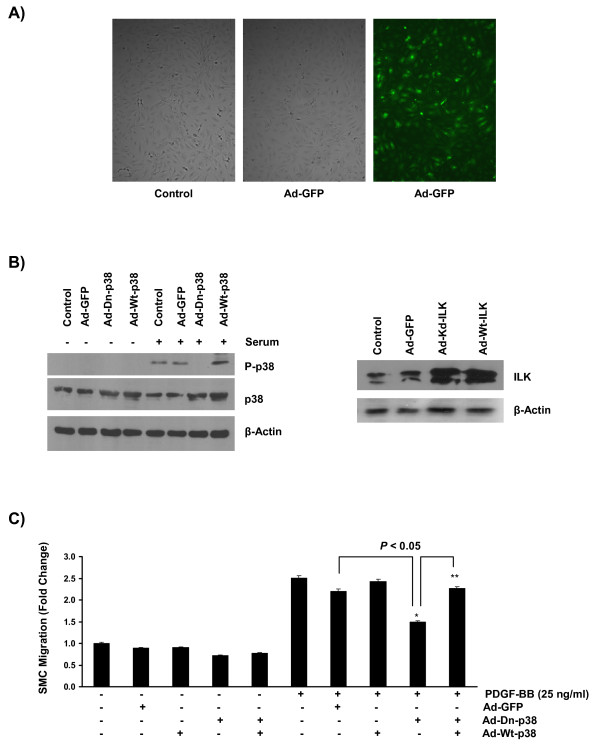
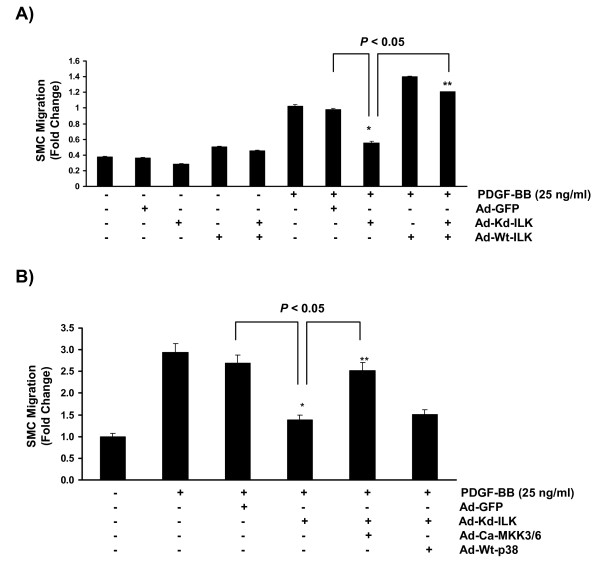
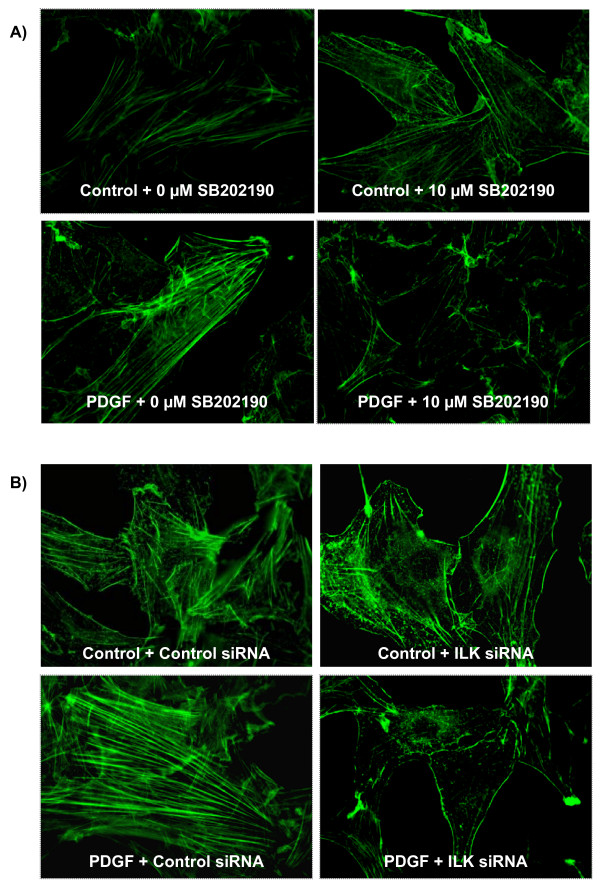
Results: In the present study, we report that integrin-linked kinase controls mouse aortic smooth muscle cell migration in response to platelet-derived growth factor. We have also identified p38 mitogen activated protein kinase as a downstream signaling pathway of the integrin-linked kinase that regulates platelet-derived growth factor-induced actin polymerization and smooth muscle cell migration.
Conclusion: This study will provide new insights into the potential therapeutic value of modulating integrin signaling in an attempt to block or delay smooth muscle cell migration and the progression of vascular diseases.
Figures
Similar articles
-
Integrin linked kinase (ILK) expression and function in vascular smooth muscle cells.Cell Adh Migr. 2009 Apr-Jun;3(2):174-6. doi: 10.4161/cam.3.2.7374. Epub 2009 Apr 10. Cell Adh Migr. 2009. PMID: 19262169 Free PMC article. Review.
-
Platelet-Derived Growth Factor Receptor-β Regulates Vascular Smooth Muscle Cell Phenotypic Transformation and Neuroinflammation After Intracerebral Hemorrhage in Mice.Crit Care Med. 2016 Jun;44(6):e390-402. doi: 10.1097/CCM.0000000000001425. Crit Care Med. 2016. PMID: 26646459 Free PMC article.
-
Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration.Circ Res. 2004 May 14;94(9):1219-26. doi: 10.1161/01.RES.0000126848.54740.4A. Epub 2004 Apr 1. Circ Res. 2004. PMID: 15059930
-
Signal transduction in matrix contraction and the migration of vascular smooth muscle cells in three-dimensional matrix.J Vasc Res. 2003 Jul-Aug;40(4):378-88. doi: 10.1159/000072702. Epub 2003 Jul 29. J Vasc Res. 2003. PMID: 12891007
-
Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes.J Cell Biol. 2001 Nov 12;155(4):505-10. doi: 10.1083/jcb.200108077. Epub 2001 Nov 5. J Cell Biol. 2001. PMID: 11696562 Free PMC article. Review.
Cited by
-
Deletion of the SPARC acidic domain or EGF-like module reduces SPARC-induced migration and signaling through p38 MAPK/HSP27 in glioma.Carcinogenesis. 2012 Feb;33(2):275-84. doi: 10.1093/carcin/bgr276. Epub 2011 Nov 23. Carcinogenesis. 2012. PMID: 22114076 Free PMC article.
-
Differential roles of CXCL2 and CXCL3 and their receptors in regulating normal and asthmatic airway smooth muscle cell migration.J Immunol. 2013 Sep 1;191(5):2731-41. doi: 10.4049/jimmunol.1203421. Epub 2013 Jul 31. J Immunol. 2013. PMID: 23904157 Free PMC article.
-
Rsu1 contributes to cell adhesion and spreading in MCF10A cells via effects on P38 map kinase signaling.Cell Adh Migr. 2015;9(3):227-32. doi: 10.4161/19336918.2014.972775. Epub 2014 Oct 23. Cell Adh Migr. 2015. PMID: 25482629 Free PMC article.
-
Molecular signatures of differential responses to exercise trainings during rehabilitation.Biomed Genet Genom. 2017;2(1):10.15761/BGG.1000127. doi: 10.15761/BGG.1000127. Epub 2017 Apr 10. Biomed Genet Genom. 2017. PMID: 28845464 Free PMC article.
-
Slug, a Cancer-Related Transcription Factor, is Involved in Vascular Smooth Muscle Cell Transdifferentiation Induced by Platelet-Derived Growth Factor-BB During Atherosclerosis.J Am Heart Assoc. 2020 Jan 21;9(2):e014276. doi: 10.1161/JAHA.119.014276. Epub 2020 Jan 21. J Am Heart Assoc. 2020. PMID: 31959031 Free PMC article.
References
-
- Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. Journal of Clinical Investigation. 1997;100(11 Suppl):S87–89. - PubMed
-
- Kher N, Marsh JD. Pathobiology of atherosclerosis--a brief review. Seminars in Thrombosis & Hemostasis. 2004;30(6):665–672. - PubMed
-
- Chen Y, Xu H, Liu J, Zhang C, Leutz A, Mo X. The c-Myb functions as a downstream target of PDGF-mediated survival signal in vascular smooth muscle cells. Biochemical & Biophysical Research Communications. 2007;360(2):433–436. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
