

Drugging the PI3 kinome: from chemical tools to drugs in the clinic
- PMID: 20179189
- PMCID: PMC3242038
- DOI: 10.1158/0008-5472.CAN-09-4355
Drugging the PI3 kinome: from chemical tools to drugs in the clinic
Abstract
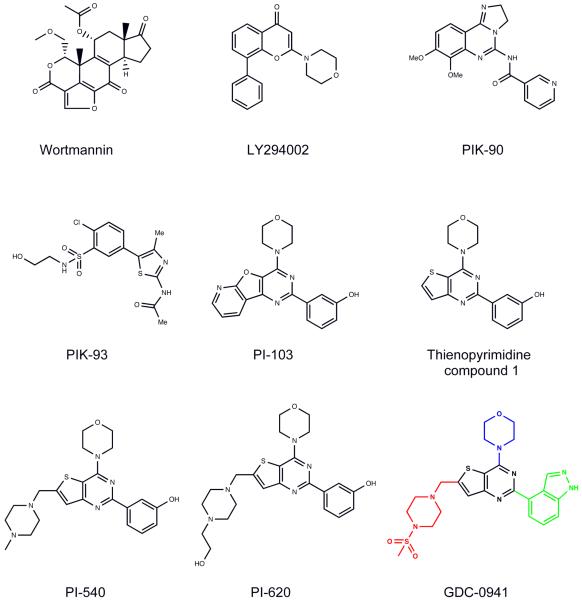
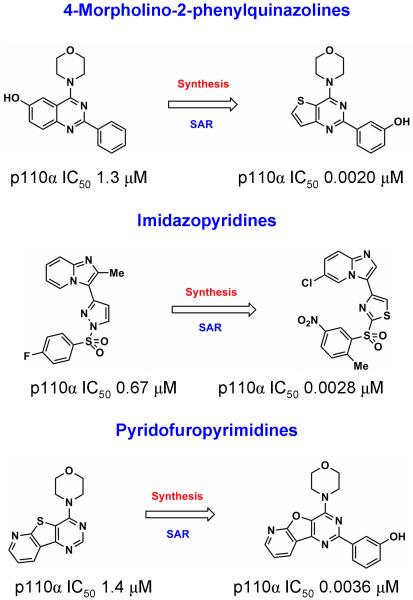
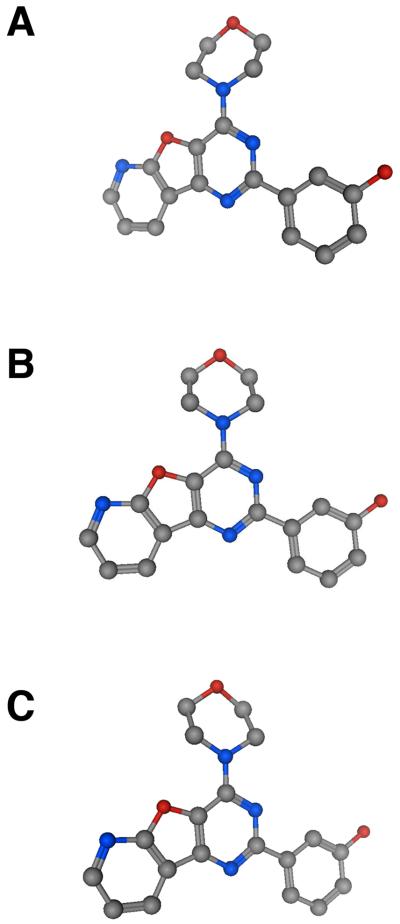
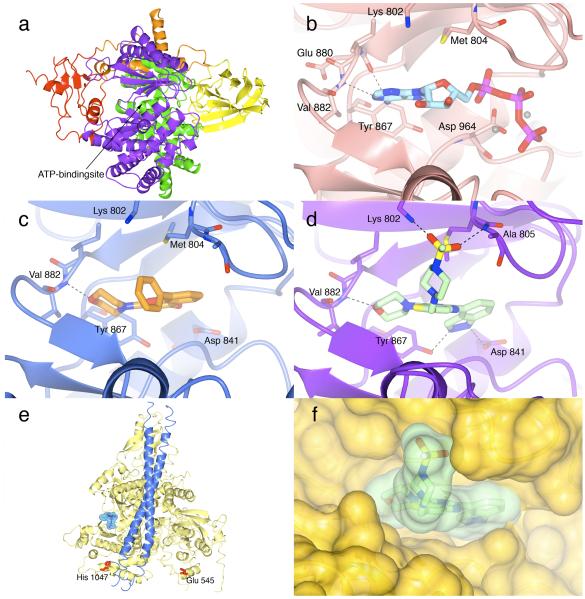
The phosphatidylinositide 3-kinase (PI3K) pathway is very commonly activated in a wide range of human cancers and is a major driving force in oncogenesis. One of the class I lipid kinase members of the PI3K family, p110alpha, is probably the most commonly mutated kinase in the human genome. Alongside genetic, molecular biological, and biochemical studies, chemical inhibitors have been extremely helpful tools in understanding the role of PI3K enzymes in signal transduction and downstream physiological and pathological processes, and also in validating PI3Ks as therapeutic targets. Although they have been valuable in the past, the early and still frequently employed inhibitors, wortmannin and LY294002, have significant limitations as chemical tools. Here, we discuss the case history of the discovery and properties of an increasingly used chemical probe, the pan-class I PI3K and mammalian target of rapamycin (mTOR) inhibitor PI-103 (a pyridofuropyrimidine), and its very recent evolution into the thienopyrimidine drug GDC-0941, which exhibits excellent oral anticancer activity in preclinical models and is now undergoing phase I clinical trials in cancer patients. We also illustrate the impact of structural biology on the design of PI3K inhibitors and on the interpretation of their effects. The challenges and outlook for drugging the PI3 kinome are discussed in the more general context of the role of structural biology and chemical biology in innovative drug discovery.
Figures
References
-
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Ann Rev Biochem. 2001;70:535–602. - PubMed
-
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. - PubMed
-
- Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
