

Lights on for aminopeptidases in cystic kidney disease
- PMID: 20179346
- PMCID: PMC2827971
- DOI: 10.1172/JCI42378
Lights on for aminopeptidases in cystic kidney disease
Abstract
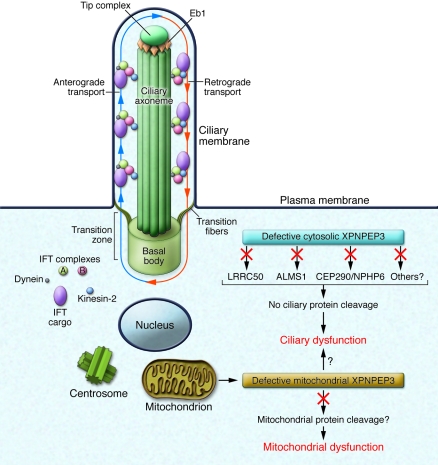
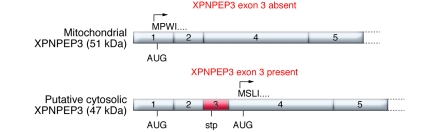
While erudite cell biologists have for many decades described singular immotile appendages known as primary cilia to be present on most cells in our bodies, cilial function(s) long remained an enigma. Driven largely by an ever increasing number of discoveries of genetic defects in primary cilia during the past decade, cilia were catapulted from a long lasting existence in obscurity into the bright spotlight in cell biology and medicine. The study by O'Toole et al. in this issue of the JCI adds a novel "enzymatic" facet to the rapidly growing information about these little cellular tails, by demonstrating that defects in the XPNPEP3 gene, which encodes mitochondrial and cytosolic splice variants of X-prolyl aminopeptidase 3, can cause nephronophthisis-like ciliopathy. Future studies are in order now to elucidate the cystogenic pathways affected by disrupted enzymatic function of XPNPEP3 in cilia-related cystogenic diseases.
Figures
Comment on
-
Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy.J Clin Invest. 2010 Mar;120(3):791-802. doi: 10.1172/JCI40076. Epub 2010 Feb 22. J Clin Invest. 2010. PMID: 20179356 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
