

Retracted: Inhibition of multiple protective signaling pathways and Ad.5/3 delivery enhances mda-7/IL-24 therapy of malignant glioma
- PMID: 20179672
- PMCID: PMC2889737
- DOI: 10.1038/mt.2010.29
Retracted: Inhibition of multiple protective signaling pathways and Ad.5/3 delivery enhances mda-7/IL-24 therapy of malignant glioma
Retraction in
-
Retraction Notice to: Inhibition of Multiple Protective Signaling Pathways and Ad.5/3 Delivery Enhances mda-7/IL-24 Therapy of Malignant Glioma.Mol Ther. 2024 Dec 4;32(12):4524. doi: 10.1016/j.ymthe.2024.08.020. Epub 2024 Aug 29. Mol Ther. 2024. PMID: 39214081 Free PMC article. No abstract available.
Abstract
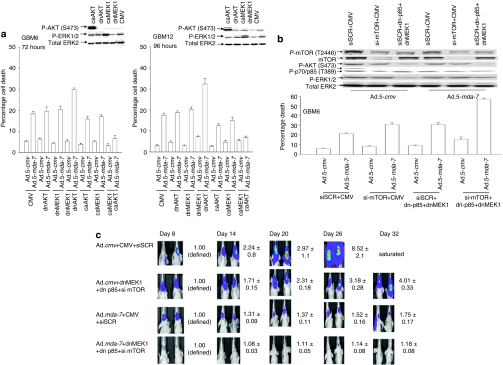
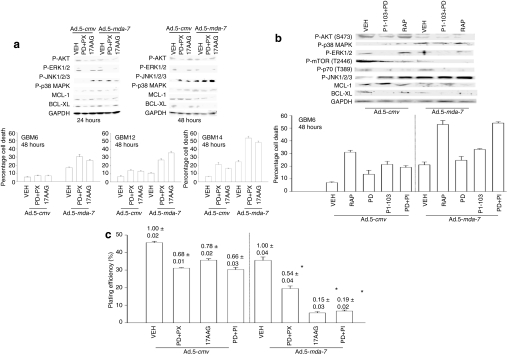
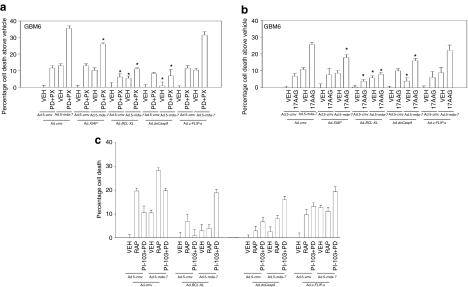
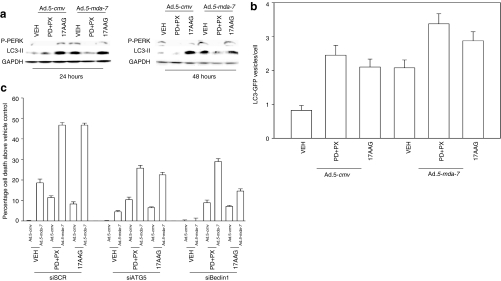
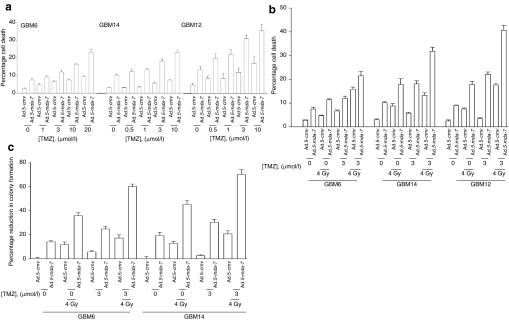
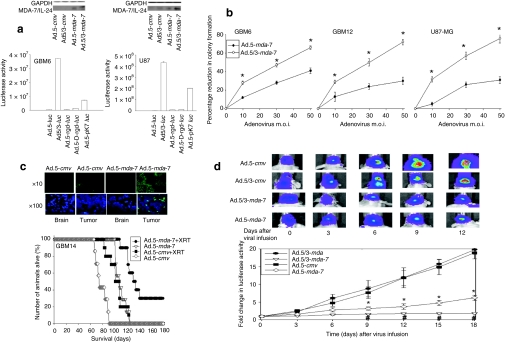
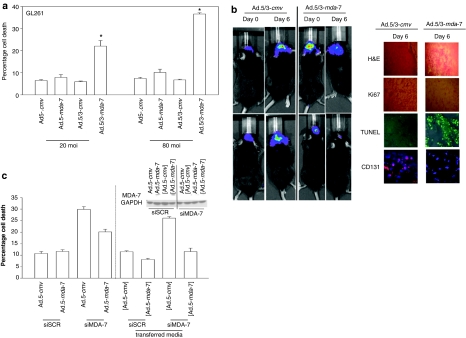
We have explored the mechanism by which inhibition of multiple cytoprotective cell-signaling pathways enhance melanoma differentiation-associated gene-7/interleukin-24 (mda-7/IL-24) toxicity toward invasive primary human glioblastoma multiforme (GBM) cells, and whether improving adenoviral infectivity/delivery of mda-7/IL-24 enhances therapeutic outcome in animals containing orthotopic xenografted GBM cells. The toxicity of a serotype 5 recombinant adenovirus to express MDA-7/IL-24 (Ad.5-mda-7) was enhanced by combined molecular or small molecule inhibition of mitogen-activated extracellular regulated kinase (MEK)1/2 and phosphatidyl inositol 3-kinase (PI3K) or AKT; inhibition of mammalian target of rapamycin (mTOR) and MEK1/2; and the HSP90 inhibitor 17AAG. Molecular inhibition of mTOR/PI3K/MEK1 signaling in vivo also enhanced Ad.5-mda-7 toxicity. In GBM cells of diverse genetic backgrounds, inhibition of cytoprotective cell-signaling pathways enhanced MDA-7/IL-24-induced autophagy, mitochondrial dysfunction and tumor cell death. Due partly to insufficient adenovirus serotype 5 gene delivery this therapeutic approach has shown limited success in GBM. To address this problem, we employed a recombinant adenovirus that comprises the tail and shaft domains of a serotype 5 virus and the knob domain of a serotype 3 virus expressing MDA-7/IL-24, Ad.5/3-mda-7. Ad.5/3-mda-7 more effectively infected and killed GBM cells in vitro and in vivo than Ad.5-mda-7. Future combinations of these approaches hold promise for developing an effective therapy for GBM.
Figures
References
-
- Robins HI, Chang S, Butowski N., and , Mehta M. Therapeutic advances for glioblastoma multiforme: current status and future prospects. Curr Oncol Rep. 2007;9:66–70. - PubMed
-
- Jiang H, Lin JJ, Su ZZ, Goldstein NI., and , Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995;11:2477–2486. - PubMed
-
- Ekmekcioglu S, Ellerhorst J, Mhashilkar AM, Sahin AA, Read CM, Prieto VG, et al. Down-regulated melanoma differentiation associated gene (mda-7) expression in human melanomas. Int J Cancer. 2001;94:54–59. - PubMed
-
- Ellerhorst JA, Prieto VG, Ekmekcioglu S, Broemeling L, Yekell S, Chada S, et al. Loss of MDA-7 expression with progression of melanoma. J Clin Oncol. 2002;20:1069–1074. - PubMed
-
- Huang EY, Madireddi MT, Gopalkrishnan RV, Leszczyniecka M, Su Z, Lebedeva IV, et al. Genomic structure, chromosomal localization and expression profile of a novel melanoma differentiation associated (mda-7) gene with cancer specific growth suppressing and apoptosis inducing properties. Oncogene. 2001;20:7051–7063. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01-NS031492/NS/NINDS NIH HHS/United States
- R01 CA127641/CA/NCI NIH HHS/United States
- R01 CA134721/CA/NCI NIH HHS/United States
- R01 CA063753/CA/NCI NIH HHS/United States
- R01CA63753/CA/NCI NIH HHS/United States
- R01-DK52825/DK/NIDDK NIH HHS/United States
- R01-CA134721/CA/NCI NIH HHS/United States
- R01CA77141/CA/NCI NIH HHS/United States
- R01-CA108325/CA/NCI NIH HHS/United States
- P01 CA104177/CA/NCI NIH HHS/United States
- P01-CA104177/CA/NCI NIH HHS/United States
- P01 NS031492/NS/NINDS NIH HHS/United States
- R01 CA122930/CA/NCI NIH HHS/United States
- R01 CA097318/CA/NCI NIH HHS/United States
- R01 DK052825/DK/NIDDK NIH HHS/United States
- R01-CA097318/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
