

LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma
- PMID: 20179738
- PMCID: PMC2987369
- DOI: 10.1038/ejhg.2010.11
LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma
Abstract
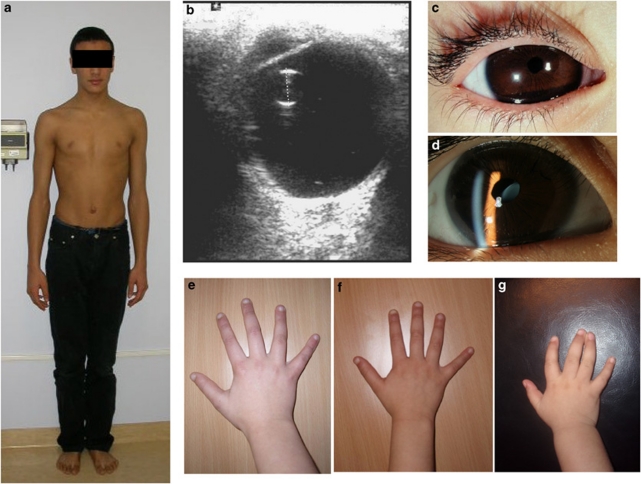
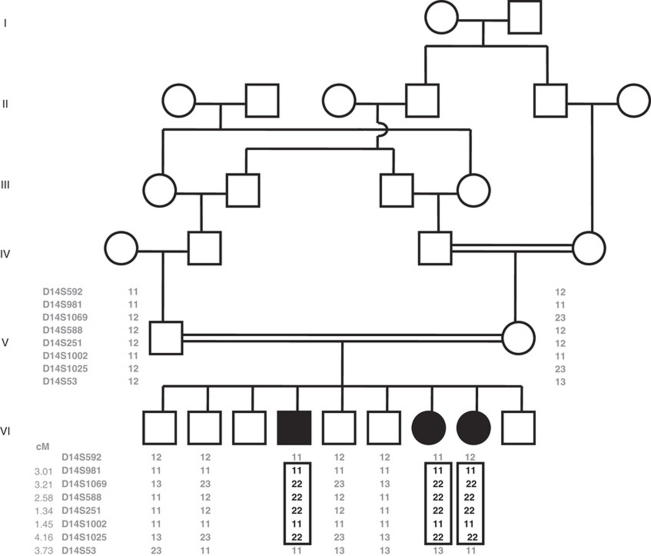
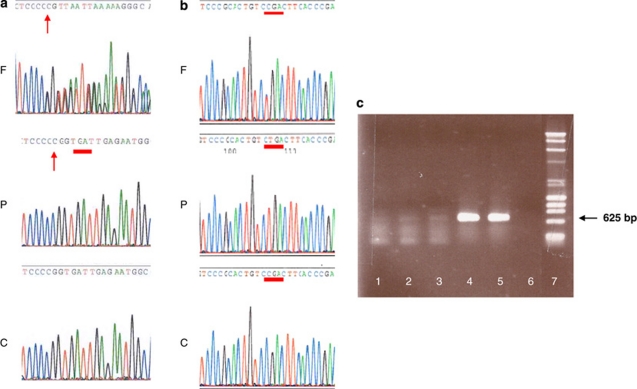
The latent TGFbeta-binding proteins (LTBPs) and fibrillins are a superfamily of large, multidomain proteins with structural and TGFbeta-signalling roles in the extracellular matrix. Their importance is underscored by fibrillin-1 mutations responsible for Marfan syndrome, but their respective roles are still incompletely understood. We report here on two families where children from healthy, consanguineous parents, presented with megalocornea and impaired vision associated with small, round, dislocated lenses (microspherophakia and ectopia lentis) and myopia, as well as a high-arched palate, and, in older children, tall stature with an abnormally large arm span over body height ratio, that is, associated features of Marfan syndrome. Glaucoma was not present at birth, but was diagnosed in older children. Whole genome homozygosity mapping followed by candidate gene analysis identified homozygous truncating mutations of LTBP2 gene in patients from both families. Fibroblast mRNA analysis was consistent with nonsense-mediated mRNA decay, with no evidence of mutated exon skipping. We conclude that biallelic null LTBP2 mutations cause the ocular phenotype in both families and could lead to Marfan-like features in older children. We suggest that intraocular pressures should be followed-up in young children with an ocular phenotype consisting of megalocornea, spherophakia and/or lens dislocation, and recommend LTBP2 gene analysis in these patients.
Figures
References
-
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. - PubMed
-
- Faivre L, Masurel-Paulet A, Collod-Beroud G, et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123:391–398. - PubMed
-
- Meire FM, Delleman WJ, Bleeker-Wagemakers EM. Ocular manifestations of congenital Marfan syndrome with contractures (CMC syndrome) Ophthalmic Paediatr Genet. 1991;12:1–9. - PubMed
-
- Meire FM, Van EJ, Hanssens M. Congenital Marfan syndrome with contractures. A clinicopathological report. Bull Soc Belge Ophtalmol. 1992;245:91–97. - PubMed
-
- Ramirez F, Dietz HC. Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J Cell Physiol. 2007;213:326–330. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
