

Mechanistic issues of the interaction of the hairpin-forming domain of tBid with mitochondrial cardiolipin
- PMID: 20179769
- PMCID: PMC2825271
- DOI: 10.1371/journal.pone.0009342
Mechanistic issues of the interaction of the hairpin-forming domain of tBid with mitochondrial cardiolipin
Abstract
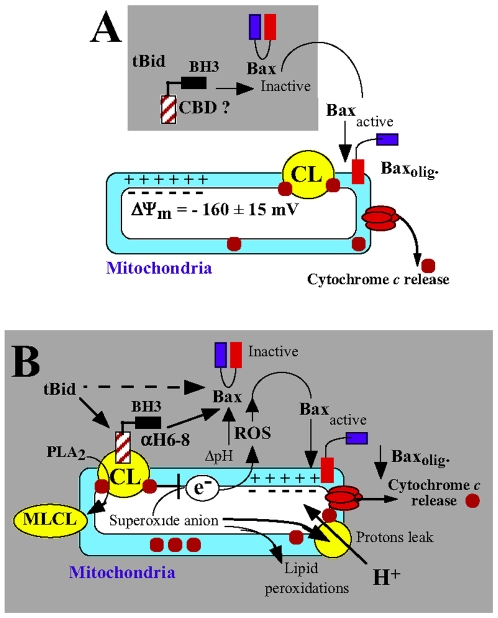
Background: The pro-apoptotic effector Bid induces mitochondrial apoptosis in synergy with Bax and Bak. In response to death receptors activation, Bid is cleaved by caspase-8 into its active form, tBid (truncated Bid), which then translocates to the mitochondria to trigger cytochrome c release and subsequent apoptosis. Accumulating evidence now indicate that the binding of tBid initiates an ordered sequences of events that prime mitochondria from the action of Bax and Bak: (1) tBid interacts with mitochondria via a specific binding to cardiolipin (CL) and immediately disturbs mitochondrial structure and function idependently of its BH3 domain; (2) Then, tBid activates through its BH3 domain Bax and/or Bak and induces their subsequent oligomerization in mitochondrial membranes. To date, the underlying mechanism responsible for targeting tBid to mitochondria and disrupting mitochondrial bioenergetics has yet be elucidated.
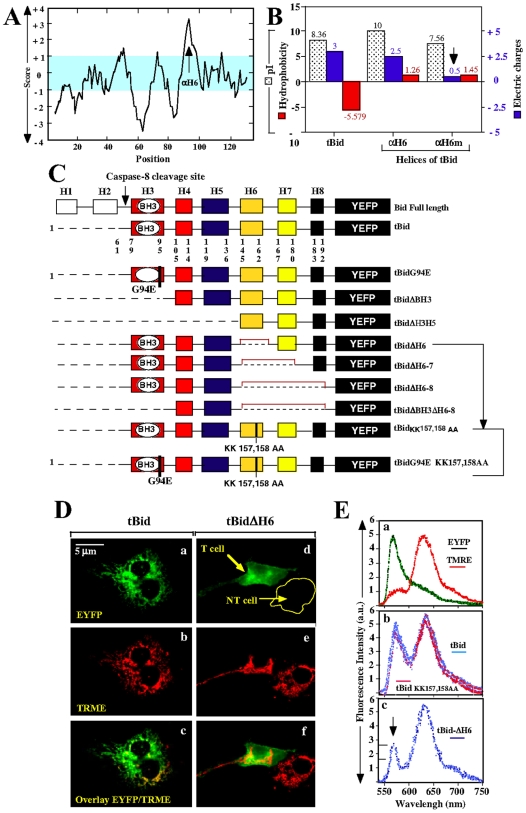
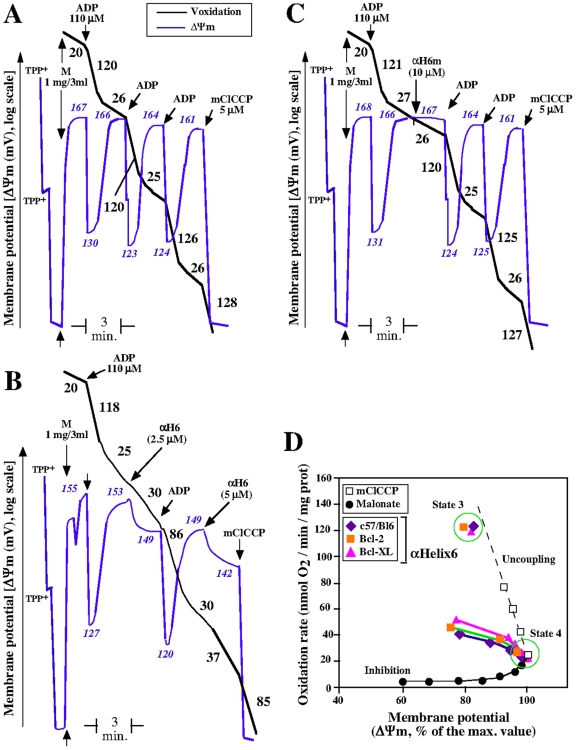
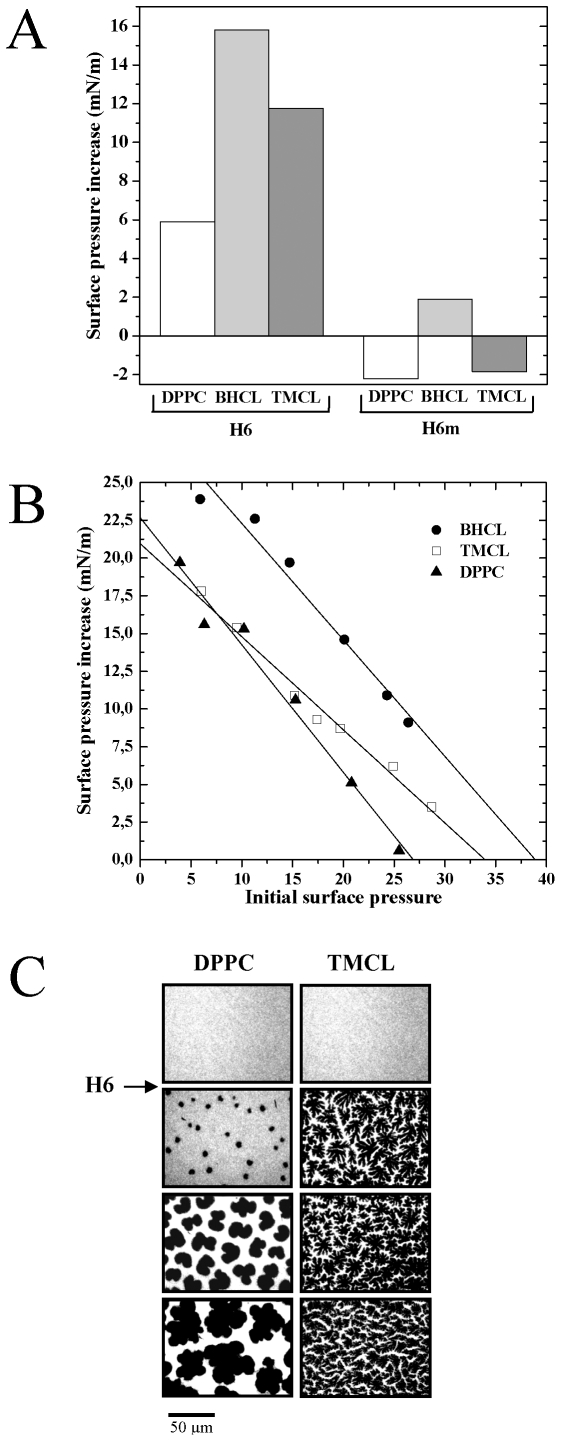
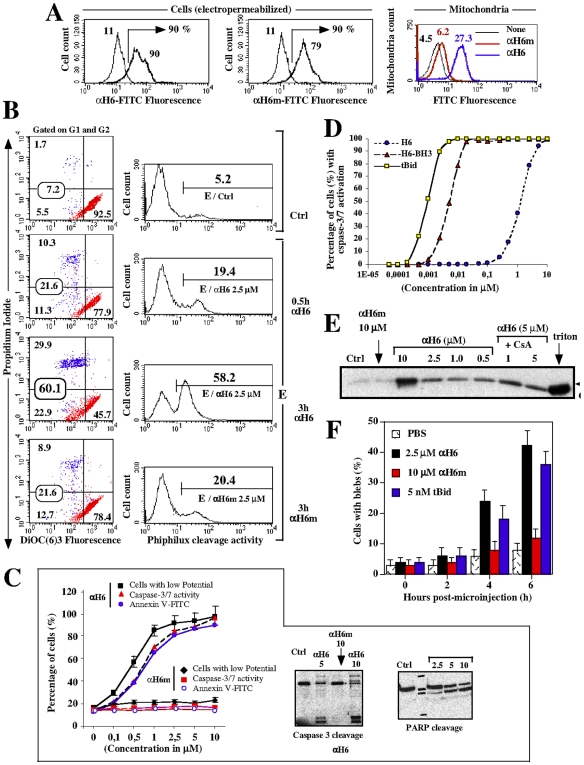
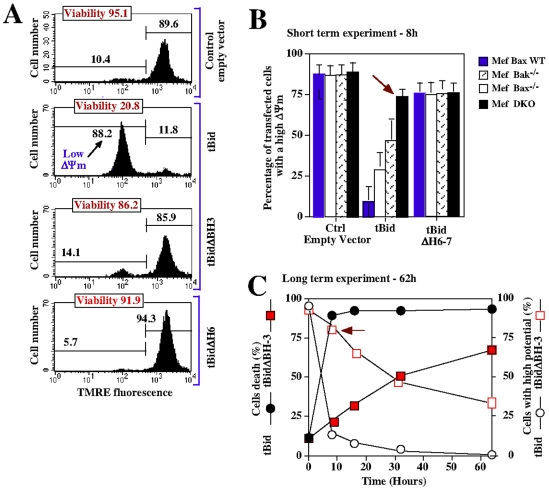
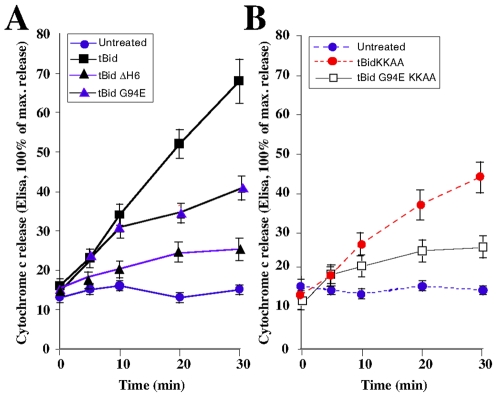
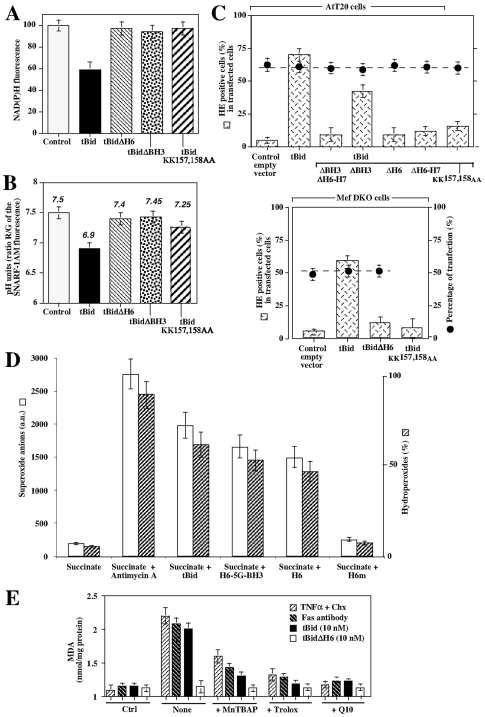
Principal findings: The present study investigates the mechanism by which tBid interacts with mitochondria issued from mouse hepatocytes and perturbs mitochondrial function. We show here that the helix alphaH6 is responsible for targeting tBid to mitochondrial CL and disrupting mitochondrial bioenergetics. In particular, alphaH6 interacts with mitochondria through electrostatic interactions involving the lysines 157 and 158 and induces an inhibition of state-3 respiration and an uncoupling of state-4 respiration. These changes may represent a key event that primes mitochondria for the action of Bax and Bak. In addition, we also demonstrate that tBid required its helix alphaH6 to efficiently induce cytochrome c release and apoptosis.
Conclusions: Our findings provide new insights into the mechanism of action of tBid, and particularly emphasize the importance of the interaction of the helix alphaH6 with CL for both mitochondrial targeting and pro-apoptotic activity of tBid. These support the notion that tBid acts as a bifunctional molecule: first, it binds to mitochondrial CL via its helix alphaH6 and destabilizes mitochondrial structure and function, and then it promotes through its BH3 domain the activation and oligomerization of Bax and/or Bak, leading to cytochrome c release and execution of apoptosis. Our findings also imply an active role of the membrane in modulating the interactions between Bcl-2 proteins that has so far been underestimated.
Conflict of interest statement
Figures
References
-
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. - PubMed
-
- Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. - PubMed
-
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, et al. X-ray and NMR structure of human Bcl-xL, and inhibitor of programmed cell death. Nature. 1996;381:335–341. - PubMed
-
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. - PubMed
-
- Letai A, Bassik M, Walensky L, Sorcinelli M, Weiler S, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
