

Reconstructing genome trees of prokaryotes using overlapping genes
- PMID: 20181237
- PMCID: PMC2845580
- DOI: 10.1186/1471-2105-11-102
Reconstructing genome trees of prokaryotes using overlapping genes
Abstract
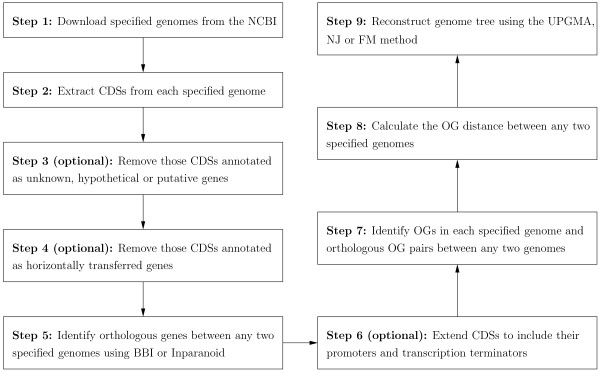
Background: Overlapping genes (OGs) are defined as adjacent genes whose coding sequences overlap partially or entirely. In fact, they are ubiquitous in microbial genomes and more conserved between species than non-overlapping genes. Based on this property, we have previously implemented a web server, named OGtree, that allows the user to reconstruct genome trees of some prokaryotes according to their pairwise OG distances. By analogy to the analyses of gene content and gene order, the OG distance between two genomes we defined was based on a measure of combining OG content (i.e., the normalized number of shared orthologous OG pairs) and OG order (i.e., the normalized OG breakpoint distance) in their whole genomes. A shortcoming of using the concept of breakpoints to define the OG distance is its inability to analyze the OG distance of multi-chromosomal genomes. In addition, the amount of overlapping coding sequences between some distantly related prokaryotic genomes may be limited so that it is hard to find enough OGs to properly evaluate their pairwise OG distances.
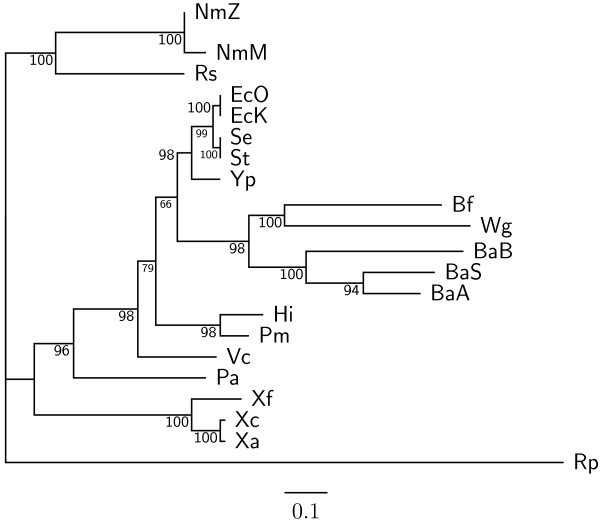
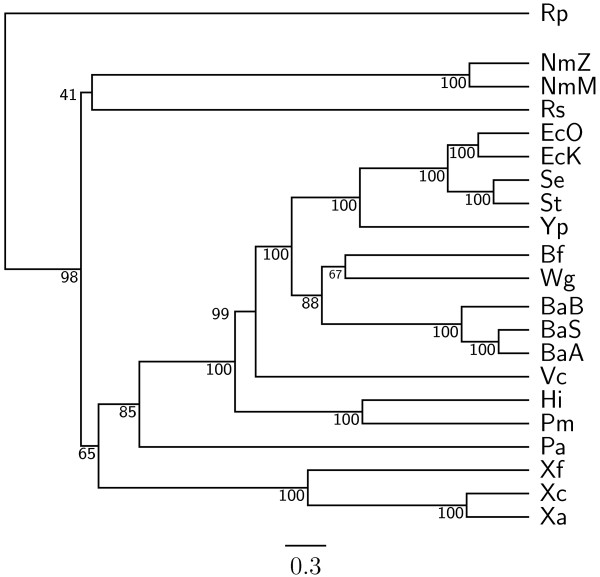
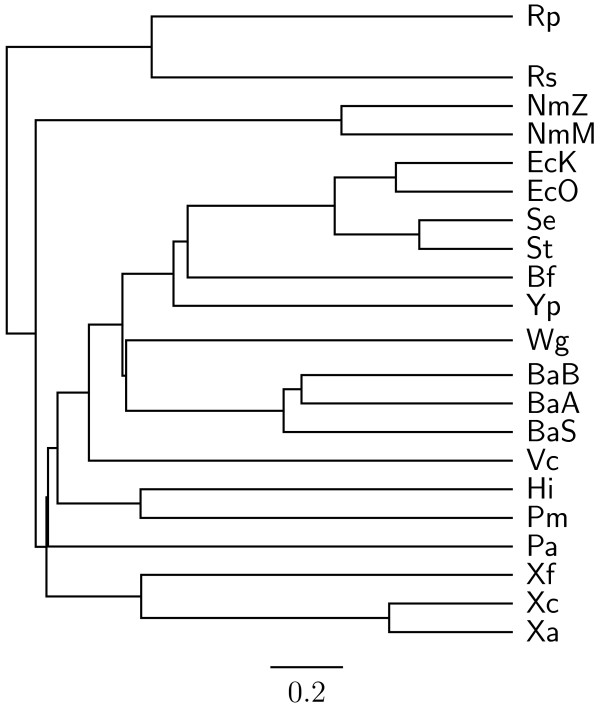
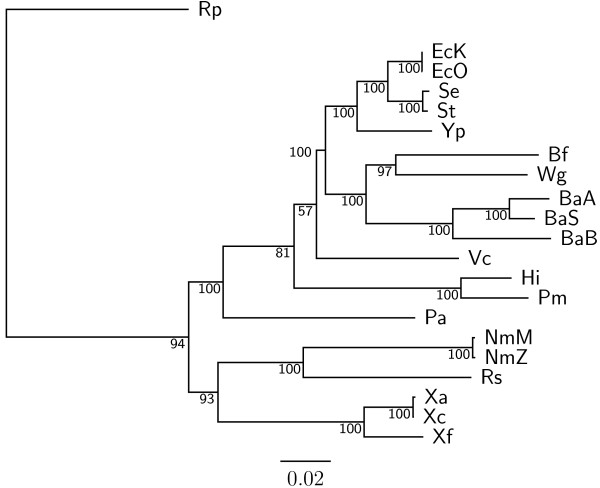
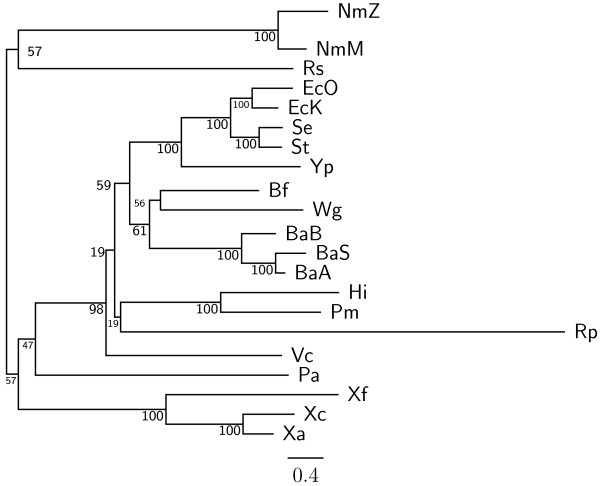
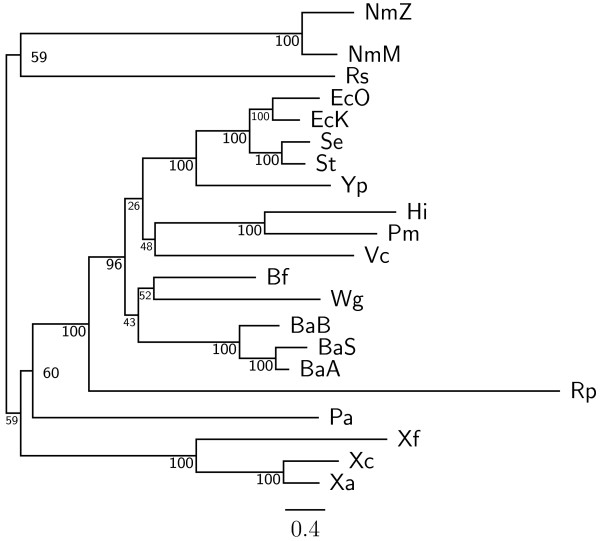
Results: In this study, we therefore define a new OG order distance that is based on more biologically accurate rearrangements (e.g., reversals, transpositions and translocations) rather than breakpoints and that is applicable to both uni-chromosomal and multi-chromosomal genomes. In addition, we expand the term "gene" to include both its coding sequence and regulatory regions so that two adjacent genes whose coding sequences or regulatory regions overlap with each other are considered as a pair of overlapping genes. This is because overlapping of regulatory regions of distinct genes suggests that the regulation of expression for these genes should be more or less interrelated. Based on these modifications, we have reimplemented our OGtree as a new web server, named OGtree2, and have also evaluated its accuracy of genome tree reconstruction on a testing dataset consisting of 21 Proteobacteria genomes. Our experimental results have finally shown that our current OGtree2 indeed outperforms its previous version OGtree, as well as another similar server, called BPhyOG, significantly in the quality of genome tree reconstruction, because the phylogenetic tree obtained by OGtree2 is greatly congruent with the reference tree that coincides with the taxonomy accepted by biologists for these Proteobacteria.
Conclusions: In this study, we have introduced a new web server OGtree2 at http://bioalgorithm.life.nctu.edu.tw/OGtree2.0/ that can serve as a useful tool for reconstructing more precise and robust genome trees of prokaryotes according to their overlapping genes.
Figures

Similar articles
-
OGtree: a tool for creating genome trees of prokaryotes based on overlapping genes.Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W475-80. doi: 10.1093/nar/gkn240. Epub 2008 May 2. Nucleic Acids Res. 2008. PMID: 18456706 Free PMC article.
-
BPhyOG: an interactive server for genome-wide inference of bacterial phylogenies based on overlapping genes.BMC Bioinformatics. 2007 Jul 25;8:266. doi: 10.1186/1471-2105-8-266. BMC Bioinformatics. 2007. PMID: 17650344 Free PMC article.
-
SoRT2: a tool for sorting genomes and reconstructing phylogenetic trees by reversals, generalized transpositions and translocations.Nucleic Acids Res. 2010 Jul;38(Web Server issue):W221-7. doi: 10.1093/nar/gkq520. Epub 2010 Jun 10. Nucleic Acids Res. 2010. PMID: 20538651 Free PMC article.
-
Genome trees and the nature of genome evolution.Annu Rev Microbiol. 2005;59:191-209. doi: 10.1146/annurev.micro.59.030804.121233. Annu Rev Microbiol. 2005. PMID: 16153168 Review.
-
Comparative Genomics for Prokaryotes.Methods Mol Biol. 2018;1704:55-78. doi: 10.1007/978-1-4939-7463-4_3. Methods Mol Biol. 2018. PMID: 29277863 Review.
Cited by
-
Exploration of multivariate analysis in microbial coding sequence modeling.BMC Bioinformatics. 2012 May 14;13:97. doi: 10.1186/1471-2105-13-97. BMC Bioinformatics. 2012. PMID: 22583558 Free PMC article.
-
Evolutionary dynamics of overlapped genes in Salmonella.PLoS One. 2013 Nov 29;8(11):e81016. doi: 10.1371/journal.pone.0081016. eCollection 2013. PLoS One. 2013. PMID: 24312259 Free PMC article.
-
Phylogeny Inference of Closely Related Bacterial Genomes: Combining the Features of Both Overlapping Genes and Collinear Genomic Regions.Evol Bioinform Online. 2015 Dec 17;11(Suppl 2):1-9. doi: 10.4137/EBO.S33491. eCollection 2015. Evol Bioinform Online. 2015. PMID: 26715828 Free PMC article.
-
Rational design of a plasmid origin that replicates efficiently in both gram-positive and gram-negative bacteria.PLoS One. 2010 Oct 8;5(10):e13244. doi: 10.1371/journal.pone.0013244. PLoS One. 2010. PMID: 20949038 Free PMC article.
-
The distinction of CPR bacteria from other bacteria based on protein family content.Nat Commun. 2019 Sep 13;10(1):4173. doi: 10.1038/s41467-019-12171-z. Nat Commun. 2019. PMID: 31519891 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
