

Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing
- PMID: 20181289
- PMCID: PMC2838848
- DOI: 10.1186/1471-2164-11-137
Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing
Abstract
Background: Cancer cells undergo massive alterations to their DNA methylation patterns that result in aberrant gene expression and malignant phenotypes. However, the mechanisms that underlie methylome changes are not well understood nor is the genomic distribution of DNA methylation changes well characterized.
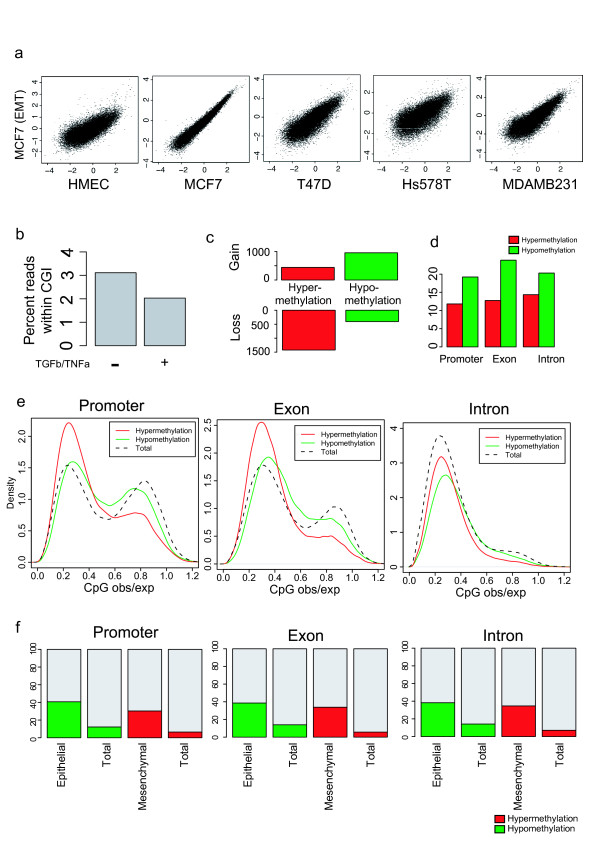
Results: Here, we performed methylated DNA immunoprecipitation combined with high-throughput sequencing (MeDIP-seq) to obtain whole-genome DNA methylation profiles for eight human breast cancer cell (BCC) lines and for normal human mammary epithelial cells (HMEC). The MeDIP-seq analysis generated non-biased DNA methylation maps by covering almost the entire genome with sufficient depth and resolution. The most prominent feature of the BCC lines compared to HMEC was a massively reduced methylation level particularly in CpG-poor regions. While hypomethylation did not appear to be associated with particular genomic features, hypermethylation preferentially occurred at CpG-rich gene-related regions independently of the distance from transcription start sites. We also investigated methylome alterations during epithelial-to-mesenchymal transition (EMT) in MCF7 cells. EMT induction was associated with specific alterations to the methylation patterns of gene-related CpG-rich regions, although overall methylation levels were not significantly altered. Moreover, approximately 40% of the epithelial cell-specific methylation patterns in gene-related regions were altered to those typical of mesenchymal cells, suggesting a cell-type specific regulation of DNA methylation.
Conclusions: This study provides the most comprehensive analysis to date of the methylome of human mammary cell lines and has produced novel insights into the mechanisms of methylome alteration during tumorigenesis and the interdependence between DNA methylome alterations and morphological changes.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
