

Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts
- PMID: 20181743
- PMCID: PMC2835323
- DOI: 10.1242/dev.049437
Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts
Abstract
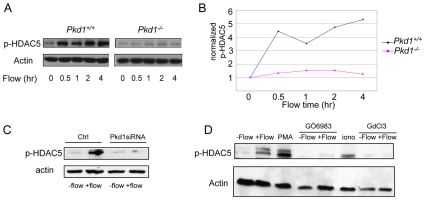
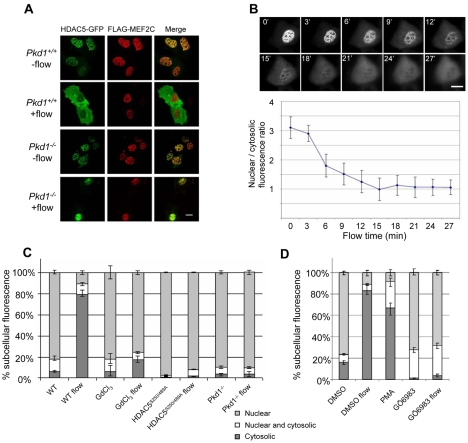
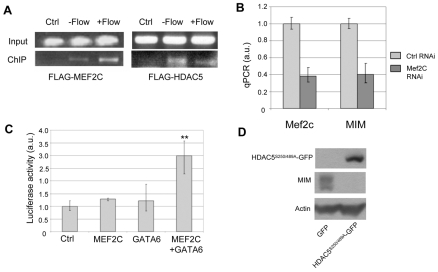
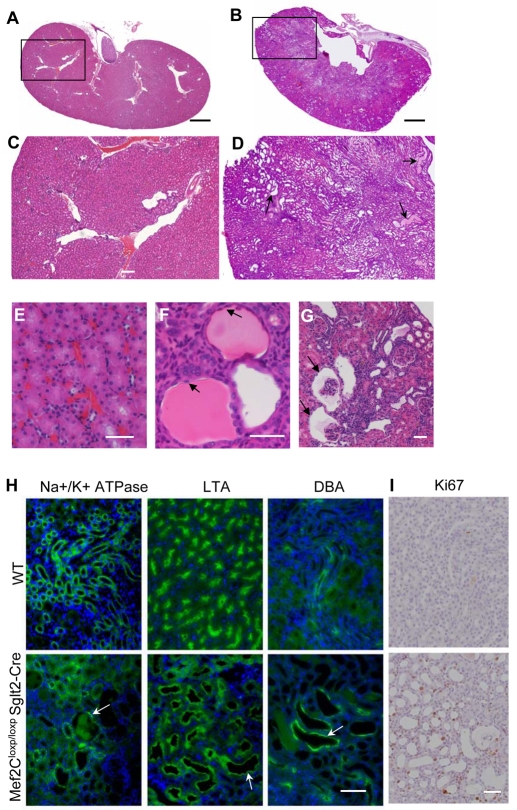
Polycystin 1 and polycystin 2 are large transmembrane proteins, which, when mutated, cause autosomal dominant polycystic kidney disease (ADPKD), a highly prevalent human genetic disease. The polycystins are thought to form a receptor-calcium channel complex in the plasma membrane of renal epithelial cells and elicit a calcium influx in response to mechanical stimulation, such as fluid flow across the apical surface of renal epithelial cells. The functional role of the polycystins in mechanosensation remains largely unknown. Here, we found that myocyte enhancer factor 2C (MEF2C) and histone deacetylase 5 (HDAC5), two key regulators of cardiac hypertrophy, are targets of polycystin-dependent fluid stress sensing in renal epithelial cells in mice. We show that fluid flow stimulation of polarized epithelial monolayers induced phosphorylation and nuclear export of HDAC5, which are crucial events in the activation of MEF2C-based transcription. Kidney-specific knockout of Mef2c, or genetrap-inactivation of a MEF2C transcriptional target, MIM, resulted in extensive renal tubule dilation and cysts, whereas Hdac5 heterozygosity or treatment with TSA, an HDAC inhibitor, reduced cyst formation in Pkd2(-/-) mouse embryos. These findings suggest a common signaling motif between myocardial hypertrophy and maintenance of renal epithelial architecture, and a potential therapeutic approach to treat ADPKD.
Figures
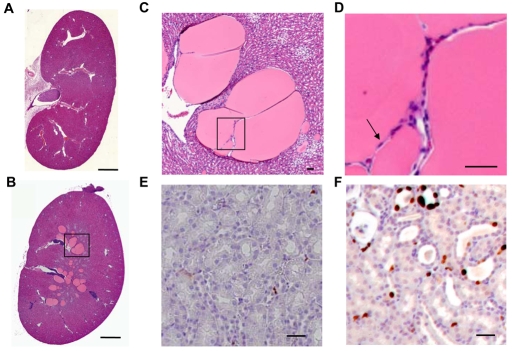
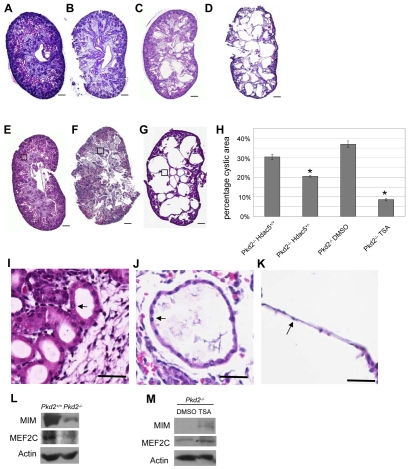
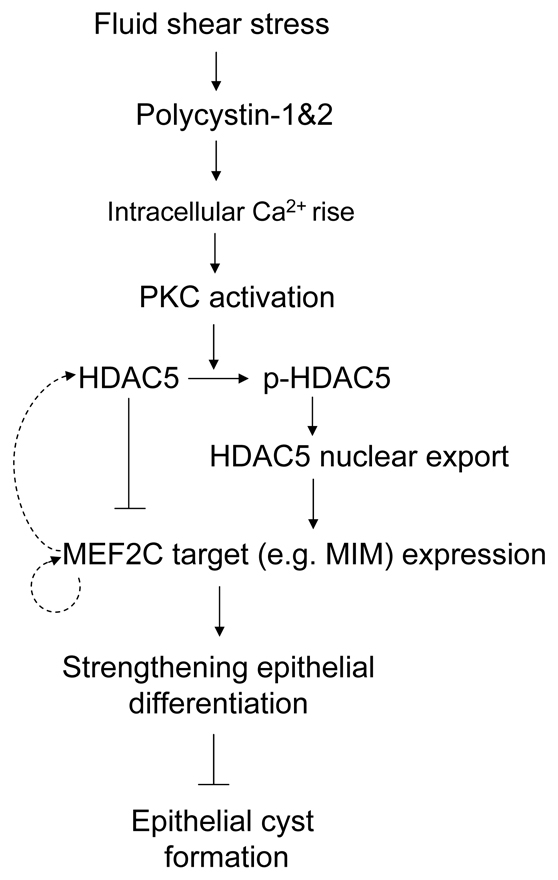
References
-
- Azzam R., Chen S. L., Shou W., Mah A. S., Alexandru G., Nasmyth K., Annan R. S., Carr S. A., Deshaies R. J. (2004). Phosphorylation by cyclin B-Cdk underlies release of mitotic exit activator Cdc14 from the nucleolus. Science 305, 516-519 - PubMed
-
- Bhunia A. K., Piontek K., Boletta A., Liu L., Qian F., Xu P. N., Germino F. J., Germino G. G. (2002). PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109, 157-168 - PubMed
-
- Boletta A., Germino G. G. (2003). Role of polycystins in renal tubulogenesis. Trends Cell Biol. 19, 484-492 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
