

Caffeine and the control of cerebral hemodynamics
- PMID: 20182032
- PMCID: PMC2944660
- DOI: 10.3233/JAD-2010-091261
Caffeine and the control of cerebral hemodynamics
Abstract
While the influence of caffeine on the regulation of brain perfusion has been the subject of multiple publications, the mechanisms involved in that regulation remain unclear. To some extent, that uncertainty is a function of a complex interplay of processes arising from multiple targets of caffeine located on a variety of different cells, many of which have influence, either directly or indirectly, on cerebral vascular smooth muscle tone. Adding to that complexity are the target-specific functional changes that may occur when comparing acute and chronic caffeine exposure. In the present review, we discuss some of the mechanisms behind caffeine influences on cerebrovascular function. The major effects of caffeine on the cerebral circulation can largely be ascribed to its inhibitory effects on adenosine receptors. Herein, we focus mostly on the A1, A2A, and A2B subtypes located in cells comprising the neurovascular unit (neurons, astrocytes, vascular smooth muscle); their roles in the coupling of increased neuronal (synaptic) activity to vasodilation; how caffeine, through blockade of these receptors, may interfere with the "neurovascular coupling" process; and receptor-linked changes that may occur in cerebrovascular regulation when comparing acute to chronic caffeine intake.
Figures
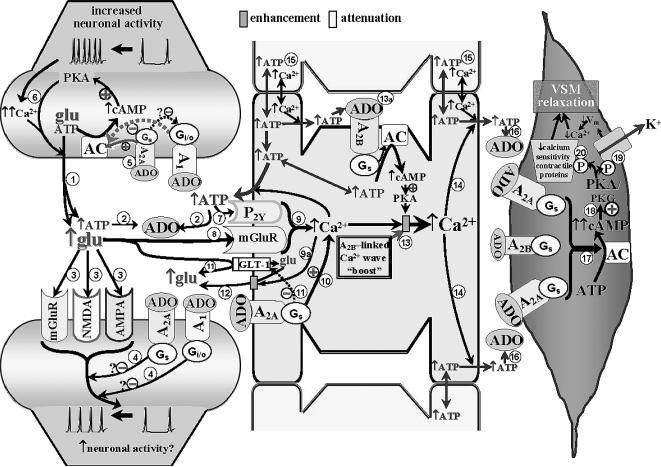
 . The released ATP can be rapidly converted to adenosine (ADO) via ectonucleotidases
. The released ATP can be rapidly converted to adenosine (ADO) via ectonucleotidases . The increased ADO can engage A1 and A2A receptors on pre- and post-synaptic membranes, and it can interact with A2A and A2B receptors on adjacent astrocytes (see below). Although A1 receptors may be expressed on astrocytes and blood vessels, in the Fig. 1 model, those sites are not assigned any functional significance (see Table 1). The glutamate released from the presynaptic terminal can effect post-synaptic activation via engaging metabotropic (mGluR) or ionotropic (NMDA and AMPA) receptors on post-synaptic dendrites
. The increased ADO can engage A1 and A2A receptors on pre- and post-synaptic membranes, and it can interact with A2A and A2B receptors on adjacent astrocytes (see below). Although A1 receptors may be expressed on astrocytes and blood vessels, in the Fig. 1 model, those sites are not assigned any functional significance (see Table 1). The glutamate released from the presynaptic terminal can effect post-synaptic activation via engaging metabotropic (mGluR) or ionotropic (NMDA and AMPA) receptors on post-synaptic dendrites . Post-synaptic activation of A1 and A2A receptors has been associated with repression of glutamate-linked post-synaptic function [22]. This could act as a “brake” on trans-synaptic signaling
. Post-synaptic activation of A1 and A2A receptors has been associated with repression of glutamate-linked post-synaptic function [22]. This could act as a “brake” on trans-synaptic signaling . The patterns of A1 and A2A receptor expression, as well as the neurotransmitters they modulate, vary among brain structures. Based upon information obtained from cerebrocortical synaptosomes (where evidence indicates the presence of both A1 and A2A receptor-mediated modulation of glutamate release [32,33]), the model depicted in Fig. 1 (and Fig. 2) could be taken to represent cerebral cortex. The figure depicts the presence of A1 and A2A receptors in close association with one another in the presynaptic nerve ending. This “heteromeric” arrangement represents one of several possibilities, including scenarios where the A1 or A2A receptor subtype predominates. In the heteromeric arrangement, it has been postulated that the Gs-linked A2A receptor not only will activate adenylyl cyclase (AC), but also, via a PKA-independent mechanism [32,33], prevent the Gi/o-linked A1 receptor from inhibiting AC, especially under conditions of increased neuronal activity and ADO availability [35]
. The patterns of A1 and A2A receptor expression, as well as the neurotransmitters they modulate, vary among brain structures. Based upon information obtained from cerebrocortical synaptosomes (where evidence indicates the presence of both A1 and A2A receptor-mediated modulation of glutamate release [32,33]), the model depicted in Fig. 1 (and Fig. 2) could be taken to represent cerebral cortex. The figure depicts the presence of A1 and A2A receptors in close association with one another in the presynaptic nerve ending. This “heteromeric” arrangement represents one of several possibilities, including scenarios where the A1 or A2A receptor subtype predominates. In the heteromeric arrangement, it has been postulated that the Gs-linked A2A receptor not only will activate adenylyl cyclase (AC), but also, via a PKA-independent mechanism [32,33], prevent the Gi/o-linked A1 receptor from inhibiting AC, especially under conditions of increased neuronal activity and ADO availability [35] . One consequence of this will be a PKA-driven increased Ca2+ influx at the presynaptic membrane, overcoming A1 receptor-linked depression of voltage-dependent Ca2+ entry [33], thereby potentiating Ca2+-dependent glutamate/ATP release and extracellular ADO generation
. One consequence of this will be a PKA-driven increased Ca2+ influx at the presynaptic membrane, overcoming A1 receptor-linked depression of voltage-dependent Ca2+ entry [33], thereby potentiating Ca2+-dependent glutamate/ATP release and extracellular ADO generation . Astrocytic Component. The released ATP and glutamate can interact with astrocyte metabotropic P2Y receptors
. Astrocytic Component. The released ATP and glutamate can interact with astrocyte metabotropic P2Y receptors and mGluR's
and mGluR's , respectively, leading to mobilization of Ca2+ from intracellular storage sites within astrocytes
, respectively, leading to mobilization of Ca2+ from intracellular storage sites within astrocytes . In addition, the increased presence of ADO, arising from the released ATP, activates A2A receptors on astrocytes leading to cAMP/PKA-dependent mobilization of intracellular Ca2+ from cellular stores
. In addition, the increased presence of ADO, arising from the released ATP, activates A2A receptors on astrocytes leading to cAMP/PKA-dependent mobilization of intracellular Ca2+ from cellular stores . Adenosine interaction with astrocytic A2A receptors also can contribute to blockade of the astrocytic glutamate import protein, GLT-1
. Adenosine interaction with astrocytic A2A receptors also can contribute to blockade of the astrocytic glutamate import protein, GLT-1 ; and promote Ca2+-dependent
; and promote Ca2+-dependent enhancement of glutamate efflux
enhancement of glutamate efflux . This should result in further elevations in glutamate levels in the synaptic cleft, as well as contributing to the astrocytic “Ca2+ wave”. The figure also speculates that a PKA-linked “boost” to the astrocytic Ca2+ mobilization
. This should result in further elevations in glutamate levels in the synaptic cleft, as well as contributing to the astrocytic “Ca2+ wave”. The figure also speculates that a PKA-linked “boost” to the astrocytic Ca2+ mobilization may arise from ADO binding to Gs-linked A2B receptors
may arise from ADO binding to Gs-linked A2B receptors . The “wave” of Ca2+ generated by the combined influences of glutamatergic, purinergic P2Y, and adenosinergic mechanisms will ultimately promote ATP release from astrocytes, including remote sites
. The “wave” of Ca2+ generated by the combined influences of glutamatergic, purinergic P2Y, and adenosinergic mechanisms will ultimately promote ATP release from astrocytes, including remote sites . ATP represents an important signaling molecule in astrocytes. It arises from cellular glucose and O2 metabolism and can diffuse (along with Ca2+) from astrocyte to astrocyte through gap junctions
. ATP represents an important signaling molecule in astrocytes. It arises from cellular glucose and O2 metabolism and can diffuse (along with Ca2+) from astrocyte to astrocyte through gap junctions . Additionally, ATP represents perhaps the most important molecule involved in inter-astrocytic communication. Thus, Ca2+-dependent release of ATP from one astrocyte interacts with P2Y receptors on adjacent astrocytes, contributing to the spread of the Ca2+ wave. Arteriolar Component. The release of ATP in the vicinity of arterioles is likely to result in rapid formation of ADO
. Additionally, ATP represents perhaps the most important molecule involved in inter-astrocytic communication. Thus, Ca2+-dependent release of ATP from one astrocyte interacts with P2Y receptors on adjacent astrocytes, contributing to the spread of the Ca2+ wave. Arteriolar Component. The release of ATP in the vicinity of arterioles is likely to result in rapid formation of ADO and interactions with smooth muscle A2 receptors. There is little doubt that cerebral arterioles are well-endowed with A2 receptors. Both A2 subtypes are likely to be present on cerebral resistance vessels; although the literature seems to favor the A2A receptor, especially in intraparenchymal and pial arterioles [24,50]. This is reflected in the figure. Principally, A2 activation generates cAMP
and interactions with smooth muscle A2 receptors. There is little doubt that cerebral arterioles are well-endowed with A2 receptors. Both A2 subtypes are likely to be present on cerebral resistance vessels; although the literature seems to favor the A2A receptor, especially in intraparenchymal and pial arterioles [24,50]. This is reflected in the figure. Principally, A2 activation generates cAMP , which is not only capable of activating PKA, but cGMP-dependent protein kinase (PKG) as well [46,70]
, which is not only capable of activating PKA, but cGMP-dependent protein kinase (PKG) as well [46,70] . The increased kinase function is associated with phosphorylation and opening of K+ channels
. The increased kinase function is associated with phosphorylation and opening of K+ channels , leading to smooth muscle cell hyperpolarization (↓Vm). This lowers intracellular Ca2+ levels through a reduction in Ca2+ influx via voltage-operated Ca2+ channels. Elevated PKA/PKG function also is accompanied by a reduction in the Ca2+-sensitivity of contractile proteins (e.g., myosin
, leading to smooth muscle cell hyperpolarization (↓Vm). This lowers intracellular Ca2+ levels through a reduction in Ca2+ influx via voltage-operated Ca2+ channels. Elevated PKA/PKG function also is accompanied by a reduction in the Ca2+-sensitivity of contractile proteins (e.g., myosin ). The combination of reduced VSM Ca2+ levels and diminished sensitivity to Ca2+ leads to relaxation. See text for further discussion and additional citations.
). The combination of reduced VSM Ca2+ levels and diminished sensitivity to Ca2+ leads to relaxation. See text for further discussion and additional citations.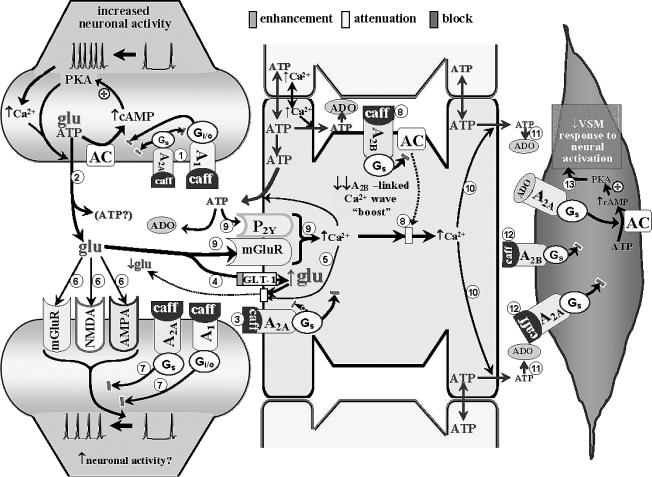
 . Although precise predictions are not possible, caffeine-related blockade of both receptors might be associated with modest or no changes in activity-evoked glutamate and ATP release
. Although precise predictions are not possible, caffeine-related blockade of both receptors might be associated with modest or no changes in activity-evoked glutamate and ATP release . Further reductions in glutamate presence in the synaptic cleft may arise from caffeine blockade of astrocytic A2A receptors
. Further reductions in glutamate presence in the synaptic cleft may arise from caffeine blockade of astrocytic A2A receptors – leading to disinhibition of GLT-1 (permitting greater uptake of glutamate from the synaptic cleft)
– leading to disinhibition of GLT-1 (permitting greater uptake of glutamate from the synaptic cleft) and diminished A2A receptor-mediated Ca2+-dependent glutamate efflux
and diminished A2A receptor-mediated Ca2+-dependent glutamate efflux . A net reduction in glutamate levels in the synaptic cleft could restrict the generation of glutamate receptor-mediated post-synaptic activation
. A net reduction in glutamate levels in the synaptic cleft could restrict the generation of glutamate receptor-mediated post-synaptic activation . Yet, the presence of caffeine will also prevent ADO-linked post-synaptic depression mediated through activation of A1 and A2A receptors
. Yet, the presence of caffeine will also prevent ADO-linked post-synaptic depression mediated through activation of A1 and A2A receptors . This may permit some post-synaptic activation to occur. Moreover, caffeine blockade of astrocytic A2A and A2B receptors
. This may permit some post-synaptic activation to occur. Moreover, caffeine blockade of astrocytic A2A and A2B receptors
 , along with the diminished contributions from metabotropic purinergic and glutamatergic receptors (arising from reduced extracellular ATP and glutamate levels
, along with the diminished contributions from metabotropic purinergic and glutamatergic receptors (arising from reduced extracellular ATP and glutamate levels ), a large reduction in the capacity to generate a Ca2+ wave might be expected
), a large reduction in the capacity to generate a Ca2+ wave might be expected , resulting in less ADO being “presented” to VSM cells
, resulting in less ADO being “presented” to VSM cells . The diminished ADO exposure, combined with caffeine blockade of VSM A receptors
. The diminished ADO exposure, combined with caffeine blockade of VSM A receptors , leaves little capacity remaining for ADO-mediated vasodilation. The potentially diminished capacity to effect “remote” increases in extracellular ADO could also interfere with heterosynaptic influences (see Fig. 1 legend); although the lack of relevant information does not permit any further discussion of this matter. The above speculation provides some (although not the only) possible explanations for the finding that acute caffeine administration profoundly attenuates the in vivo dilation of pial arterioles
, leaves little capacity remaining for ADO-mediated vasodilation. The potentially diminished capacity to effect “remote” increases in extracellular ADO could also interfere with heterosynaptic influences (see Fig. 1 legend); although the lack of relevant information does not permit any further discussion of this matter. The above speculation provides some (although not the only) possible explanations for the finding that acute caffeine administration profoundly attenuates the in vivo dilation of pial arterioles accompanying somatosensory activation in rats [54].
accompanying somatosensory activation in rats [54].Similar articles
-
A1 and A2A adenosine receptors and A1 mRNA in mouse brain: effect of long-term caffeine treatment.Brain Res. 1997 Jul 11;762(1-2):153-64. doi: 10.1016/s0006-8993(97)00378-8. Brain Res. 1997. PMID: 9262169
-
Antagonism of adenosine receptors by caffeine and caffeine metabolites in equine forebrain tissues.Am J Vet Res. 2003 Feb;64(2):216-24. doi: 10.2460/ajvr.2003.64.216. Am J Vet Res. 2003. PMID: 12602592
-
Cerebral ischemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist.Eur J Pharmacol. 1995 Dec 20;287(3):295-302. doi: 10.1016/0014-2999(95)00498-x. Eur J Pharmacol. 1995. PMID: 8991804 Free PMC article.
-
Role of the central ascending neurotransmitter systems in the psychostimulant effects of caffeine.J Alzheimers Dis. 2010;20 Suppl 1(Suppl 1):S35-49. doi: 10.3233/JAD-2010-1400. J Alzheimers Dis. 2010. PMID: 20182056 Free PMC article. Review.
-
Adenosine A1-A2A receptor heteromers: new targets for caffeine in the brain.Front Biosci. 2008 Jan 1;13:2391-9. doi: 10.2741/2852. Front Biosci. 2008. PMID: 17981720 Review.
Cited by
-
Studying cerebral hemodynamics and metabolism using simultaneous near-infrared spectroscopy and transcranial Doppler ultrasound: a hyperventilation and caffeine study.Physiol Rep. 2015 Apr;3(4):e12378. doi: 10.14814/phy2.12378. Physiol Rep. 2015. PMID: 25907789 Free PMC article.
-
Does acute caffeine ingestion alter brain metabolism in young adults?Neuroimage. 2015 Apr 15;110:39-47. doi: 10.1016/j.neuroimage.2015.01.046. Epub 2015 Jan 30. Neuroimage. 2015. PMID: 25644657 Free PMC article.
-
Mental Performance and Sport: Caffeine and Co-consumed Bioactive Ingredients.Sports Med. 2022 Dec;52(Suppl 1):69-90. doi: 10.1007/s40279-022-01796-8. Epub 2022 Nov 30. Sports Med. 2022. PMID: 36447122 Free PMC article. Review.
-
Caffeine differentially alters cortical hemodynamic activity during working memory: a near infrared spectroscopy study.BMC Res Notes. 2015 Oct 1;8:520. doi: 10.1186/s13104-015-1491-3. BMC Res Notes. 2015. PMID: 26427367 Free PMC article.
-
Caffeine increases the temporal variability of resting-state BOLD connectivity in the motor cortex.Neuroimage. 2012 Feb 1;59(3):2994-3002. doi: 10.1016/j.neuroimage.2011.10.001. Epub 2011 Oct 18. Neuroimage. 2012. PMID: 22032947 Free PMC article.
References
-
- van Gelder BM, Buijsse B, Tijhuis M, Kalmijn S, Giampaoli S, Nissinen A, Kromhout D. Coffee consumption is inversely associated with cognitive decline in elderly European men: the FINE Study. Eur J Clin Nutr. 2007;61:226–232. - PubMed
-
- Benarroch EE. Neurovascular unit dysfunction: a vascular component of Alzheimer disease? Neurology. 2007;68:1730–1732. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
