

Diseases of the nuclear envelope
- PMID: 20182615
- PMCID: PMC2828284
- DOI: 10.1101/cshperspect.a000760
Diseases of the nuclear envelope
Abstract
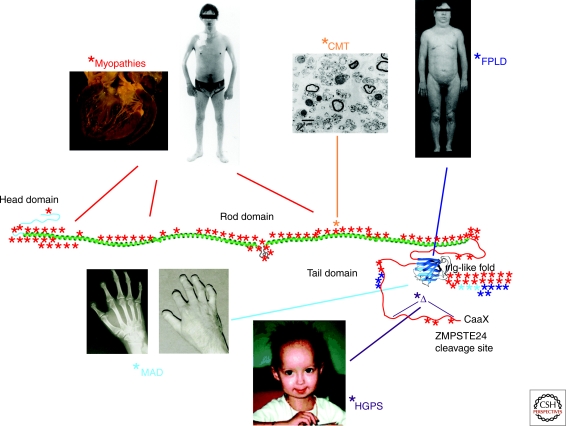
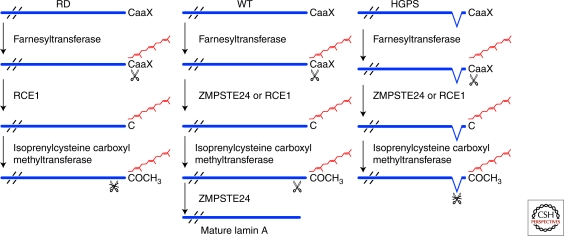
In the past decade, a wide range of fascinating monogenic diseases have been linked to mutations in the LMNA gene, which encodes the A-type nuclear lamins, intermediate filament proteins of the nuclear envelope. These diseases include dilated cardiomyopathy with variable muscular dystrophy, Dunnigan-type familial partial lipodystrophy, a Charcot-Marie-Tooth type 2 disease, mandibuloacral dysplasia, and Hutchinson-Gilford progeria syndrome. Several diseases are also caused by mutations in genes encoding B-type lamins and proteins that associate with the nuclear lamina. Studies of these so-called laminopathies or nuclear envelopathies, some of which phenocopy common human disorders, are providing clues about functions of the nuclear envelope and insights into disease pathogenesis and human aging.
Figures
Similar articles
-
The empowerment of translational research: lessons from laminopathies.Orphanet J Rare Dis. 2012 Jun 12;7:37. doi: 10.1186/1750-1172-7-37. Orphanet J Rare Dis. 2012. PMID: 22691392 Free PMC article.
-
Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies.Nucleus. 2014 Sep-Oct;5(5):427-40. doi: 10.4161/nucl.36289. Nucleus. 2014. PMID: 25482195 Free PMC article. Review.
-
The nuclear lamina and its functions in the nucleus.Int Rev Cytol. 2003;226:1-62. doi: 10.1016/s0074-7696(03)01001-5. Int Rev Cytol. 2003. PMID: 12921235 Review.
-
Towards delineating the chain of events that cause premature senescence in the accelerated aging syndrome Hutchinson-Gilford progeria (HGPS).Biochem Soc Trans. 2020 Jun 30;48(3):981-991. doi: 10.1042/BST20190882. Biochem Soc Trans. 2020. PMID: 32539085 Free PMC article. Review.
-
Laminopathies and atherosclerosis.Arterioscler Thromb Vasc Biol. 2004 Sep;24(9):1591-5. doi: 10.1161/01.ATV.0000136392.59656.8b. Epub 2004 Jun 17. Arterioscler Thromb Vasc Biol. 2004. PMID: 15205220 Review.
Cited by
-
Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport.J Cell Biol. 2013 May 13;201(4):541-57. doi: 10.1083/jcb.201212117. Epub 2013 May 6. J Cell Biol. 2013. PMID: 23649804 Free PMC article.
-
Lamin B1 loss is a senescence-associated biomarker.Mol Biol Cell. 2012 Jun;23(11):2066-75. doi: 10.1091/mbc.E11-10-0884. Epub 2012 Apr 11. Mol Biol Cell. 2012. PMID: 22496421 Free PMC article.
-
Case report: Focal segmental glomerulosclerosis in a pediatric atypical progeroid syndrome.Front Pediatr. 2022 Oct 31;10:1032653. doi: 10.3389/fped.2022.1032653. eCollection 2022. Front Pediatr. 2022. PMID: 36389384 Free PMC article.
-
The functional importance of lamins, actin, myosin, spectrin and the LINC complex in DNA repair.Exp Biol Med (Maywood). 2019 Nov;244(15):1382-1406. doi: 10.1177/1535370219876651. Epub 2019 Oct 4. Exp Biol Med (Maywood). 2019. PMID: 31581813 Free PMC article. Review.
-
Skeletal Muscle Dystrophy mutant of lamin A alters the structure and dynamics of the Ig fold domain.Sci Rep. 2018 Sep 14;8(1):13793. doi: 10.1038/s41598-018-32227-2. Sci Rep. 2018. PMID: 30218058 Free PMC article.
References
-
- Agarwal AK, Fryns JP, Auchus RJ, Garg A 2003. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 12:1995–2001 - PubMed
-
- Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L 2002. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J Am Coll Cardiol 39:981–990 - PubMed
-
- Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, Glick B, Nevo Y, Ben-Neriah Z, Melki J 2009. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet 18:3462–3469 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous