

Structural investigation of zymogenic and activated forms of human blood coagulation factor VIII: a computational molecular dynamics study
- PMID: 20184747
- PMCID: PMC2837666
- DOI: 10.1186/1472-6807-10-7
Structural investigation of zymogenic and activated forms of human blood coagulation factor VIII: a computational molecular dynamics study
Abstract
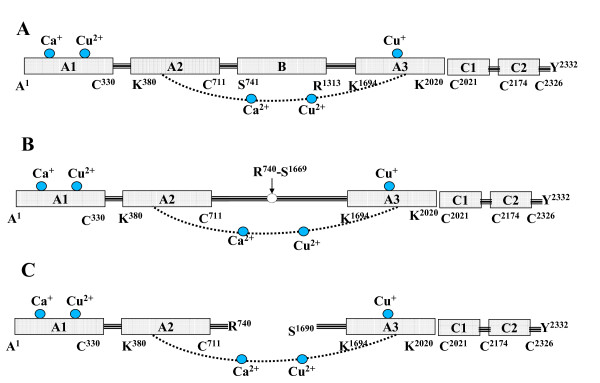
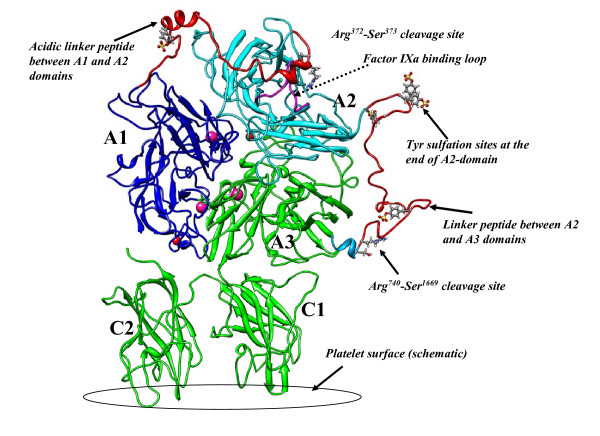
Background: Human blood coagulation factor VIII (fVIII) is a large plasma glycoprotein with sequential domain arrangement in the order A1-a1-A2-a2-B-a3-A3-C1-C2. The A1, A2 and A3 domains are interconnected by long linker peptides (a1, a2 and a3) that possess the activation sites. Proteolysis of fVIII zymogen by thrombin or factor Xa results in the generation of the activated form (fVIIIa) which serves as a critical co-factor for factor IXa (fIXa) enzyme in the intrinsic coagulation pathway.
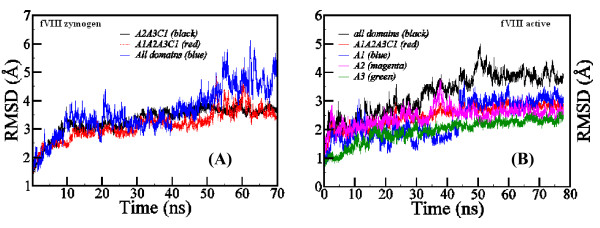
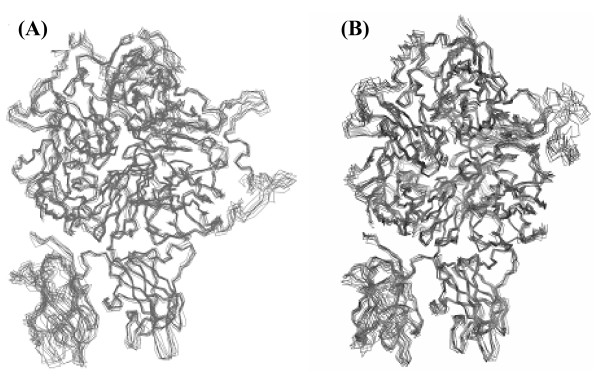
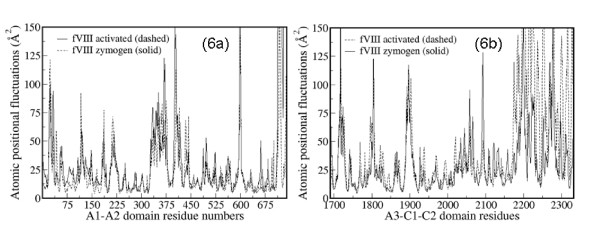
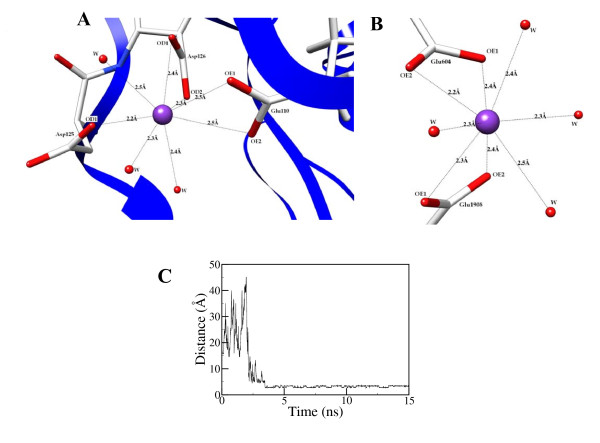
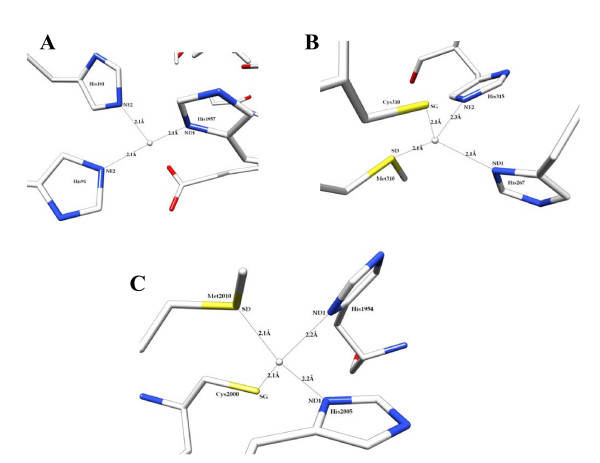
Results: In our efforts to elucidate the structural differences between fVIII and fVIIIa, we developed the solution structural models of both forms, starting from an incomplete 3.7 A X-ray crystal structure of fVIII zymogen, using explicit solvent MD simulations. The full assembly of B-domainless single-chain fVIII was built between the A1-A2 (Ala1-Arg740) and A3-C1-C2 (Ser1669-Tyr2332) domains. The structural dynamics of fVIII and fVIIIa, simulated for over 70 ns of time scale, enabled us to evaluate the integral motions of the multi-domain assembly of the co-factor and the possible coordination pattern of the functionally important calcium and copper ion binding in the protein.
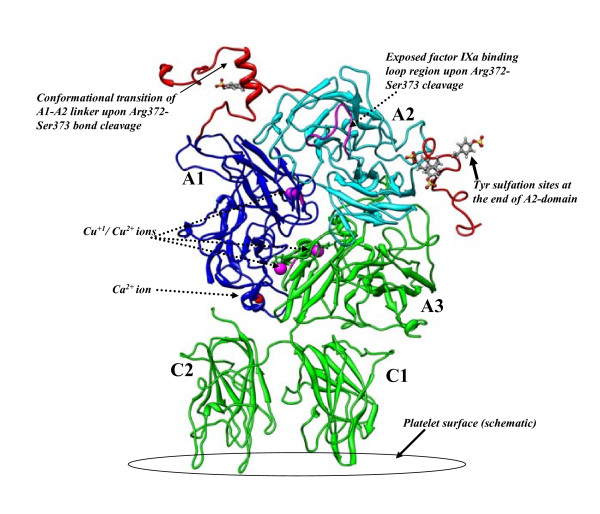
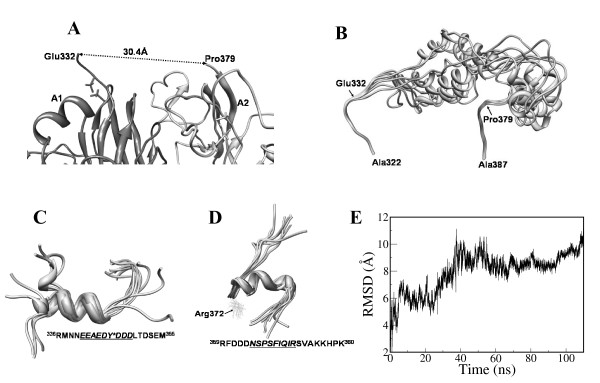
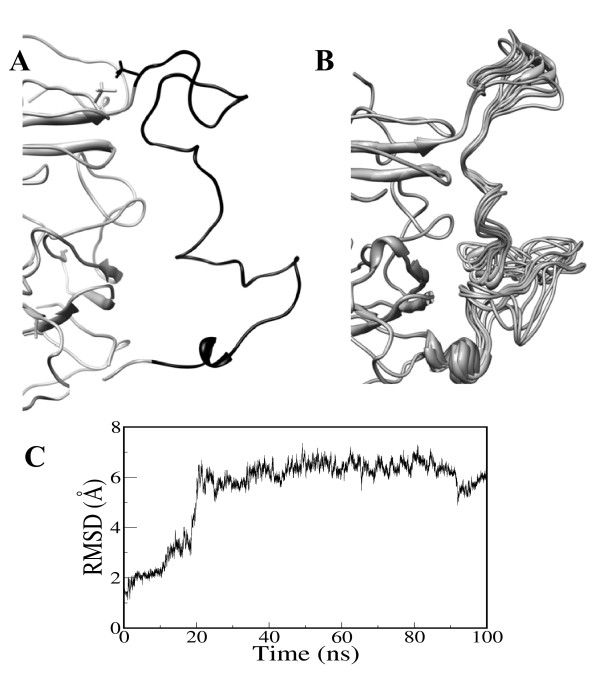
Conclusions: MD simulations predicted that the acidic linker peptide (a1) between the A1 and A2 domains is largely flexible and appears to mask the exposure of putative fIXa enzyme binding loop (Tyr555-Asp569) region in the A2 domain. The simulation of fVIIIa, generated from the zymogen structure, predicted that the linker peptide (a1) undergoes significant conformational reorganization upon activation by relocating completely to the A1-domain. The conformational transition led to the exposure of the Tyr555-Asp569 loop and the surrounding region in the A2 domain. While the proposed linker peptide conformation is predictive in nature and warrants further experimental validation, the observed conformational differences between the zymogen and activated forms may explain and support the large body of experimental data that implicated the critical importance of the cleavage of the peptide bond between the Arg372 and Ser373 residues for the full co-factor activity of fVIII.
Figures
Similar articles
-
Maturation of coagulation factor IX during Xase formation as deduced using factor VIII-derived peptides.FEBS Open Bio. 2019 Aug;9(8):1370-1378. doi: 10.1002/2211-5463.12653. Epub 2019 Jul 2. FEBS Open Bio. 2019. PMID: 31077577 Free PMC article.
-
Hydrogen-Deuterium Exchange Mass Spectrometry Identifies Activated Factor IX-Induced molecular Changes in Activated Factor VIII.Thromb Haemost. 2021 May;121(5):594-602. doi: 10.1055/s-0040-1721422. Epub 2020 Dec 10. Thromb Haemost. 2021. PMID: 33302303
-
Contribution of A1 subunit residue Q316 in thrombin-activated factor VIII to A2 subunit dissociation.Biochemistry. 2007 Aug 28;46(34):9737-42. doi: 10.1021/bi700941w. Epub 2007 Aug 4. Biochemistry. 2007. PMID: 17676877 Free PMC article.
-
Structure and function of the factor VIII gene and protein.Semin Thromb Hemost. 2003 Feb;29(1):11-22. doi: 10.1055/s-2003-37935. Semin Thromb Hemost. 2003. PMID: 12640560 Review.
-
The protein structure and effect of factor VIII.Thromb Res. 2007;119(1):1-13. doi: 10.1016/j.thromres.2005.12.015. Epub 2006 Feb 17. Thromb Res. 2007. PMID: 16487577 Review.
Cited by
-
Importance of electrostatic polarizability in calculating cysteine acidity constants and copper(I) binding energy of Bacillus subtilis CopZ.J Comput Chem. 2012 Apr 30;33(11):1142-51. doi: 10.1002/jcc.22944. Epub 2012 Feb 27. J Comput Chem. 2012. PMID: 22370900 Free PMC article.
-
Role of hydrophobic mutations on the binding affinity and stability of blood coagulation factor VIIIa: a computational molecular dynamics and free-energy analysis.Biochem Biophys Res Commun. 2014 Jul 18;450(1):735-40. doi: 10.1016/j.bbrc.2014.06.043. Epub 2014 Jun 18. Biochem Biophys Res Commun. 2014. PMID: 24952158 Free PMC article.
-
Electrostatic polarization is crucial in reproducing Cu(I) interaction energies and hydration.J Phys Chem B. 2011 Aug 25;115(33):10079-85. doi: 10.1021/jp2051933. Epub 2011 Jul 28. J Phys Chem B. 2011. PMID: 21761909 Free PMC article.
-
Structural insights into the interaction of blood coagulation co-factor VIIIa with factor IXa: a computational protein-protein docking and molecular dynamics refinement study.Biochem Biophys Res Commun. 2014 Sep 26;452(3):408-14. doi: 10.1016/j.bbrc.2014.08.078. Epub 2014 Aug 23. Biochem Biophys Res Commun. 2014. PMID: 25157807 Free PMC article.
-
Protein residue network analysis reveals fundamental properties of the human coagulation factor VIII.Sci Rep. 2021 Jun 16;11(1):12625. doi: 10.1038/s41598-021-92201-3. Sci Rep. 2021. PMID: 34135429 Free PMC article.
References
-
- Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. - PubMed
-
- van Dieijen G, Tans G, Rosing J, Hemker HC. The role of phospholipid and factor VIIIa in the activation of bovine factor X. J Biol Chem. 1981;256:3433–3442. - PubMed
-
- Fay PJ, Haidaris PJ, Smudzin TM. Human factor VIIIa subunit structure. Reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266:8957–8962. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
