

The power of protein interaction networks for associating genes with diseases
- PMID: 20185403
- PMCID: PMC2853684
- DOI: 10.1093/bioinformatics/btq076
The power of protein interaction networks for associating genes with diseases
Abstract
Motivation: Understanding the association between genetic diseases and their causal genes is an important problem concerning human health. With the recent influx of high-throughput data describing interactions between gene products, scientists have been provided a new avenue through which these associations can be inferred. Despite the recent interest in this problem, however, there is little understanding of the relative benefits and drawbacks underlying the proposed techniques.
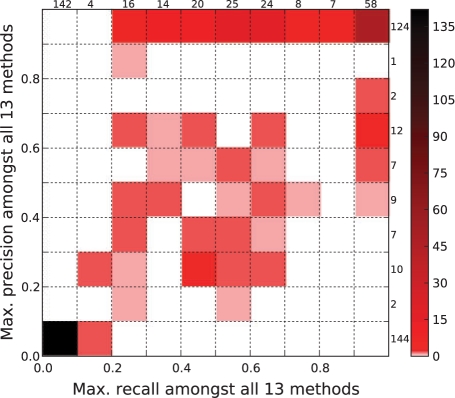
Results: We assessed the utility of physical protein interactions for determining gene-disease associations by examining the performance of seven recently developed computational methods (plus several of their variants). We found that random-walk approaches individually outperform clustering and neighborhood approaches, although most methods make predictions not made by any other method. We show how combining these methods into a consensus method yields Pareto optimal performance. We also quantified how a diffuse topological distribution of disease-related proteins negatively affects prediction quality and are thus able to identify diseases especially amenable to network-based predictions and others for which additional information sources are absolutely required.
Availability: The predictions made by each algorithm considered are available online at http://www.cbcb.umd.edu/DiseaseNet.
Figures



References
-
- Aerts S, et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. - PubMed
-
- Birnbaum S, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 2009;41:473–477. - PubMed
-
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
