

Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes
- PMID: 20185797
- PMCID: PMC2855780
- DOI: 10.1161/CIRCRESAHA.109.213116
Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes
Abstract
Rationale: NADPH oxidases are a major source of superoxide (O(2)(-)) in the cardiovascular system. The function of Nox4, a member of the Nox family of NADPH oxidases, in the heart is poorly understood.
Objective: The goal of this study was to elucidate the role of Nox4 in mediating oxidative stress and growth/death in the heart.
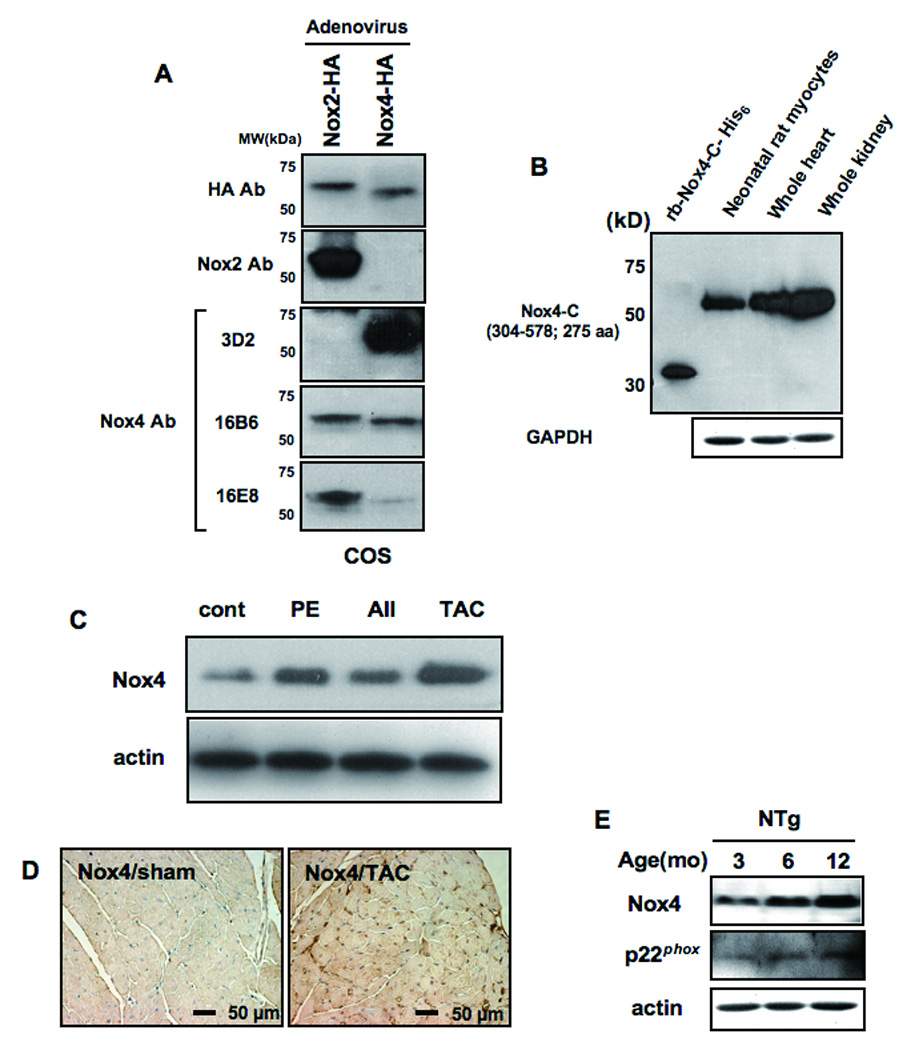
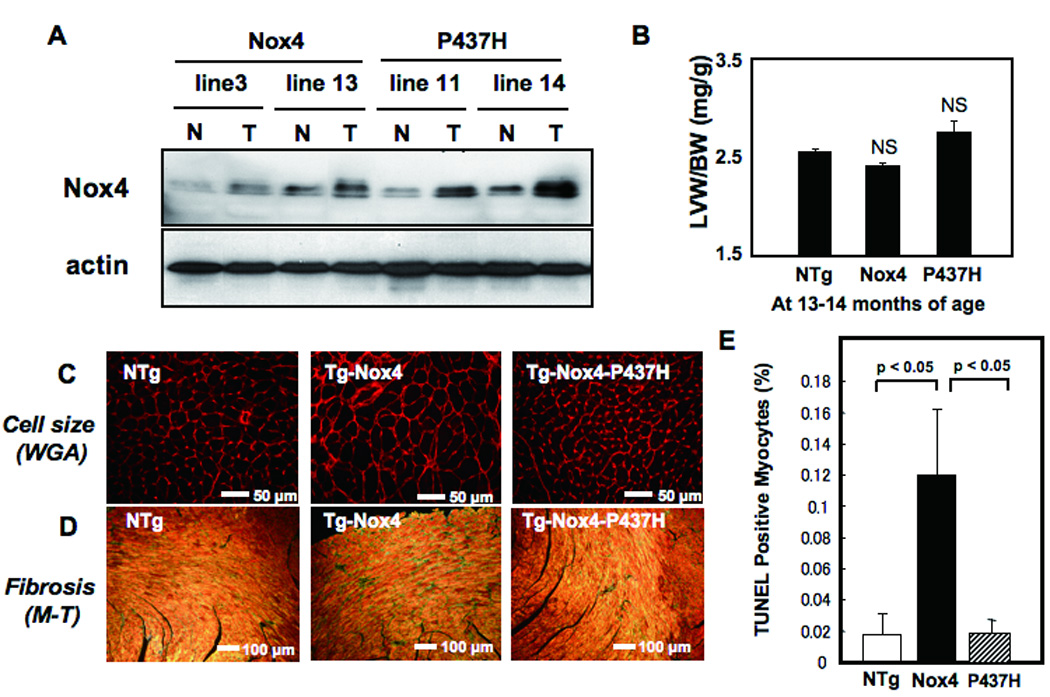
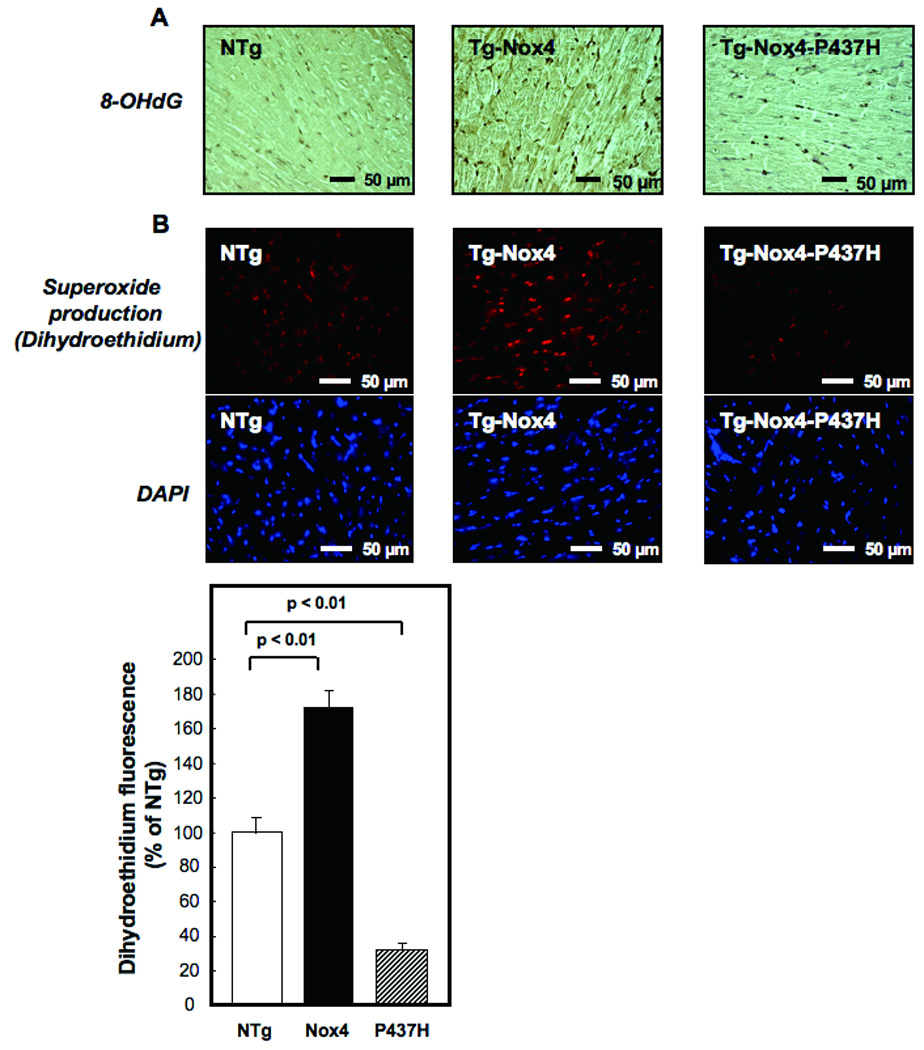
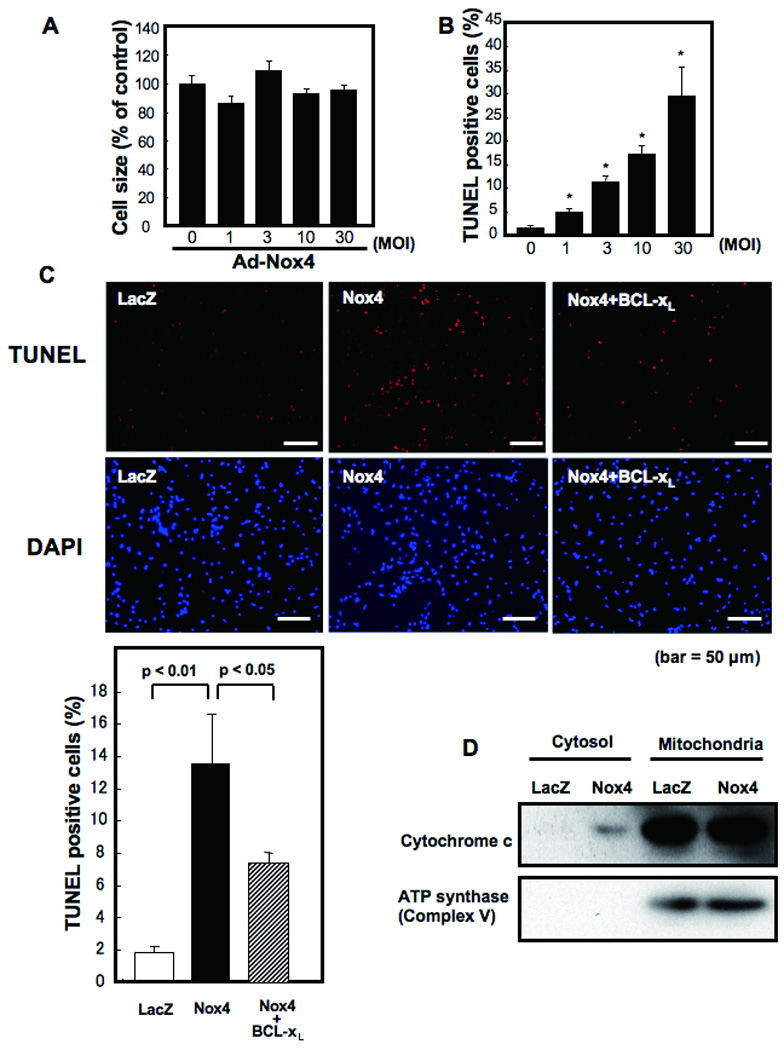
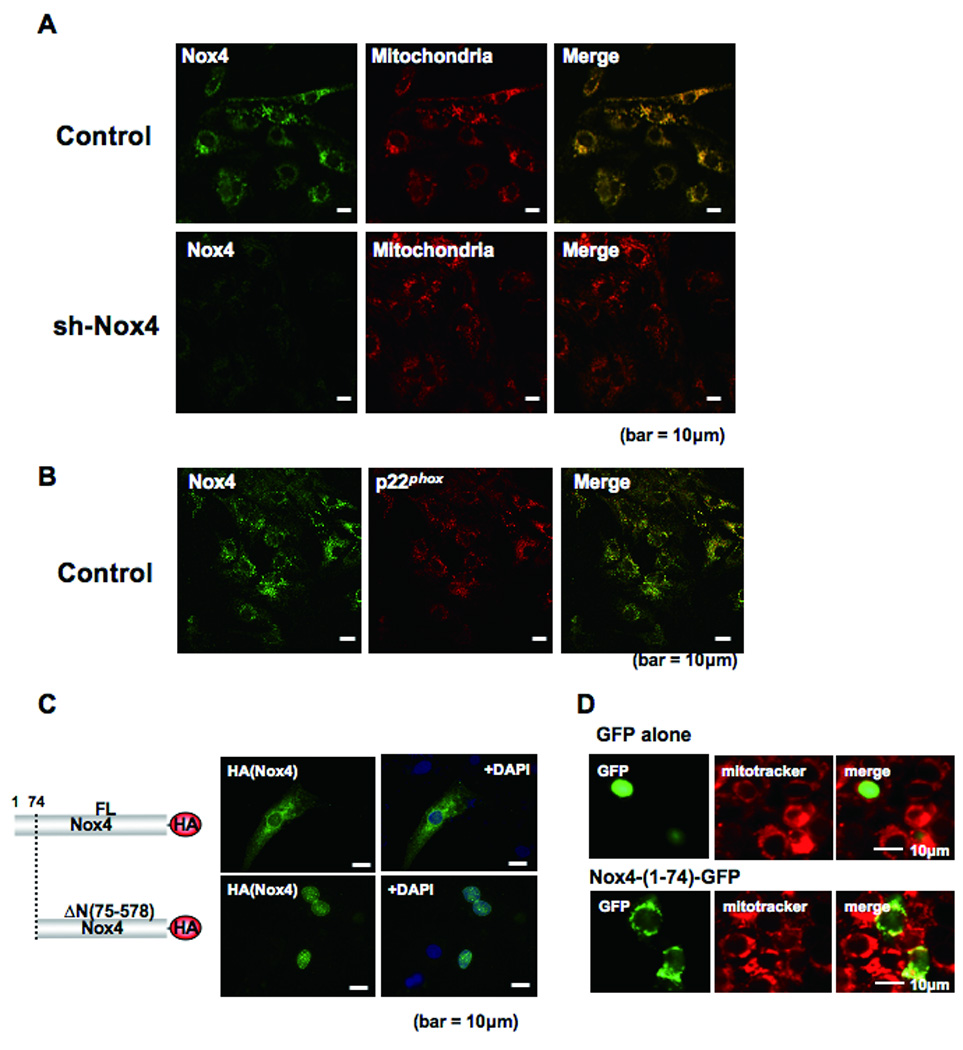
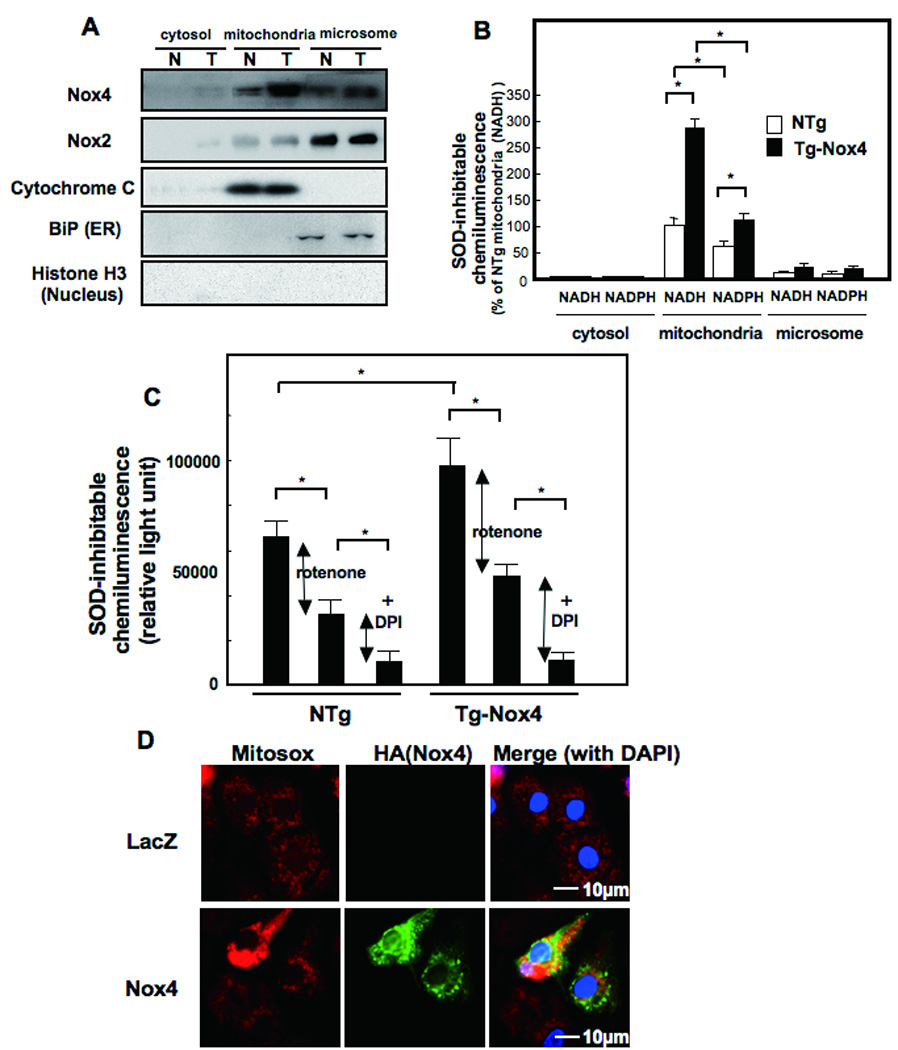
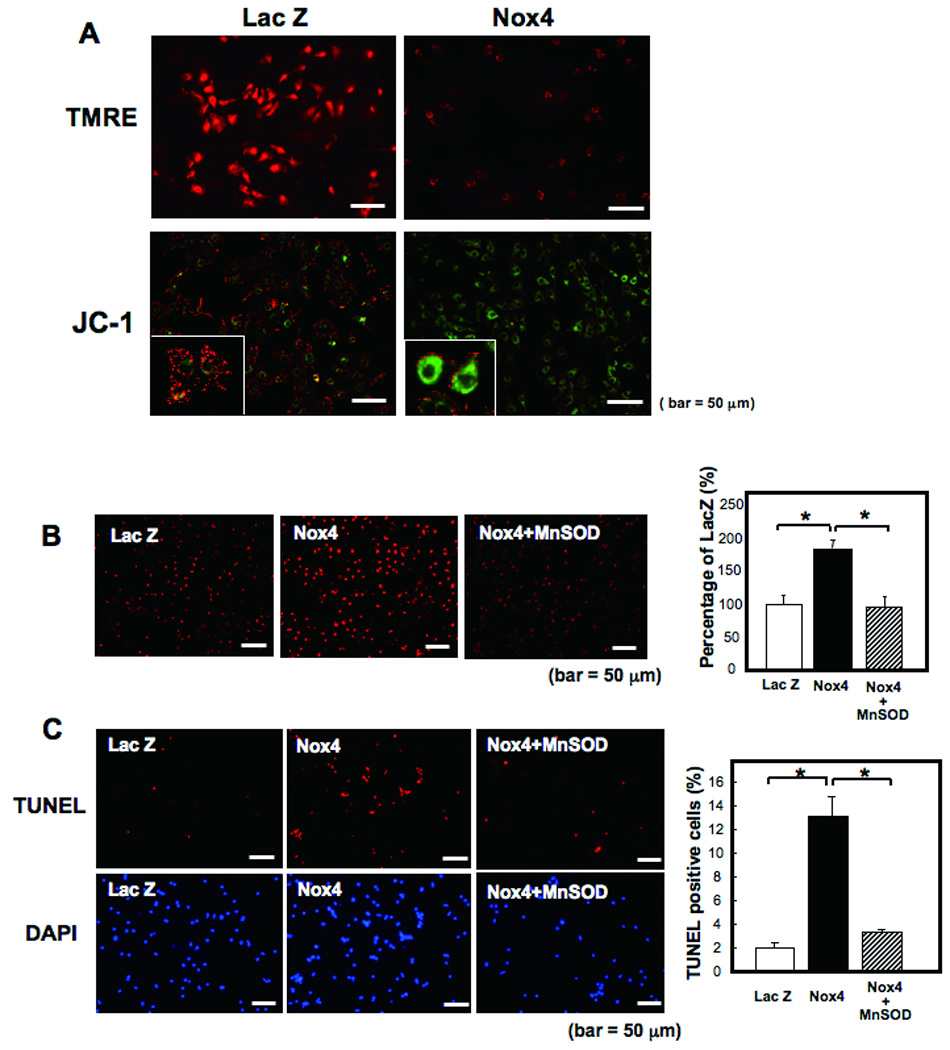
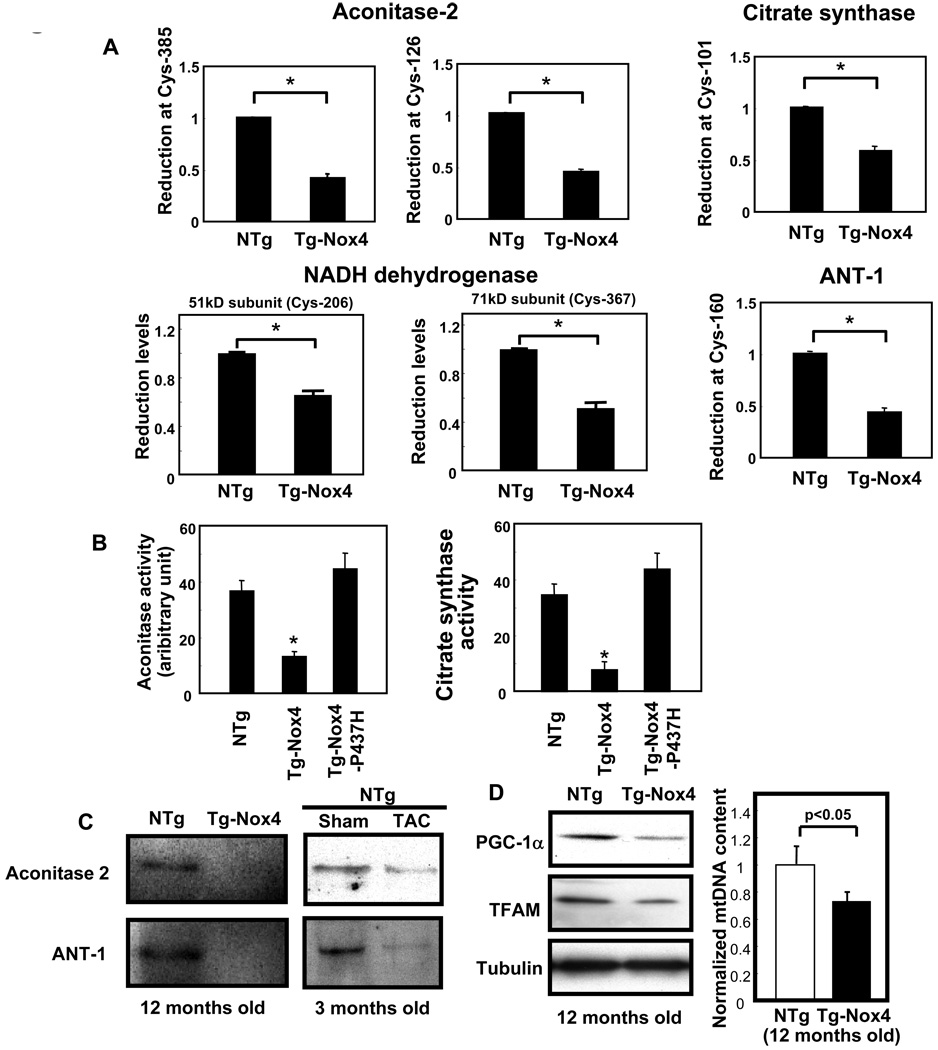
Methods and results: Expression of Nox4 in the heart was increased in response to hypertrophic stimuli and aging. Neither transgenic mice with cardiac specific overexpression of Nox4 (Tg-Nox4) nor those with catalytically inactive Nox4 (Tg-Nox4-P437H) showed an obvious baseline cardiac phenotype at young ages. Tg-Nox4 gradually displayed decreased left ventricular (LV) function with enhanced O(2)(-) production in the heart, which was accompanied by increased apoptosis and fibrosis at 13 to 14 months of age. On the other hand, the level of oxidative stress was attenuated in Tg-Nox4-P437H. Although the size of cardiac myocytes was significantly greater in Tg-Nox4 than in nontransgenic, the LV weight/tibial length was not significantly altered in Tg-Nox4 mice. Overexpression of Nox4 in cultured cardiac myocytes induced apoptotic cell death but not hypertrophy. Nox4 is primarily localized in mitochondria and upregulation of Nox4 enhanced both rotenone- and diphenyleneiodonium-sensitive O(2)(-) production in mitochondria. Cysteine residues in mitochondrial proteins, including aconitase and NADH dehydrogenases, were oxidized and their activities decreased in Tg-Nox4.
Conclusions: Upregulation of Nox4 by hypertrophic stimuli and aging induces oxidative stress, apoptosis and LV dysfunction, in part because of mitochondrial insufficiency caused by increased O(2)(-) production and consequent cysteine oxidation in mitochondrial proteins.
Figures
References
-
- Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–974. - PubMed
-
- Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002;8:132–140. - PubMed
-
- Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. - PubMed
-
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. - PubMed
-
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL091469/HL/NHLBI NIH HHS/United States
- P01 AG027211/AG/NIA NIH HHS/United States
- R01 HL033107/HL/NHLBI NIH HHS/United States
- HL69020/HL/NHLBI NIH HHS/United States
- R41 HL079729/HL/NHLBI NIH HHS/United States
- HL91469/HL/NHLBI NIH HHS/United States
- R01 HL067727/HL/NHLBI NIH HHS/United States
- P01 HL069020/HL/NHLBI NIH HHS/United States
- P01 HL059139/HL/NHLBI NIH HHS/United States
- R01 AG023039/AG/NIA NIH HHS/United States
- R01 AG028787/AG/NIA NIH HHS/United States
- HL67724/HL/NHLBI NIH HHS/United States
- AG27211/AG/NIA NIH HHS/United States
- R01 HL102738/HL/NHLBI NIH HHS/United States
- R01 HL067724/HL/NHLBI NIH HHS/United States
- HL 59139/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
