

TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor
- PMID: 20185819
- PMCID: PMC2857099
- DOI: 10.1074/jbc.M109.076091
TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor
Abstract
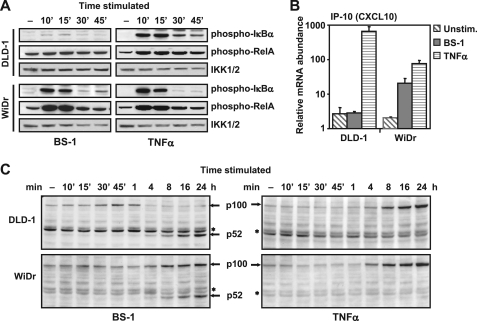
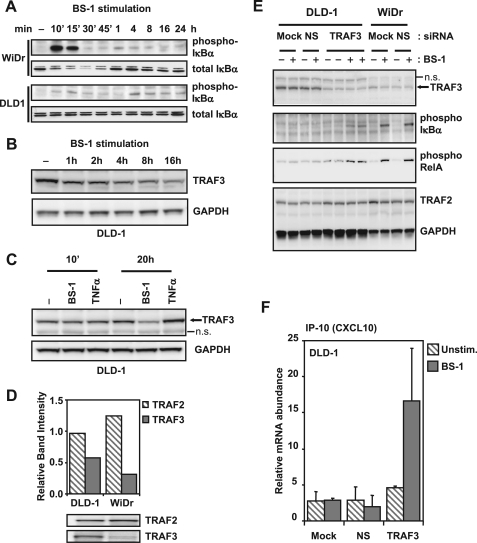
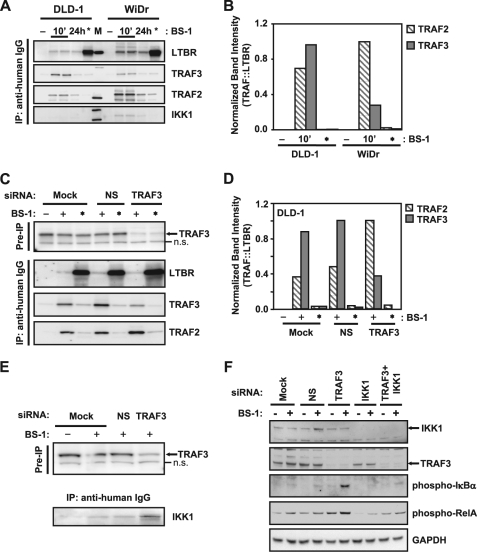
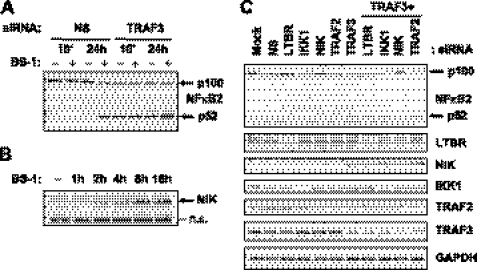
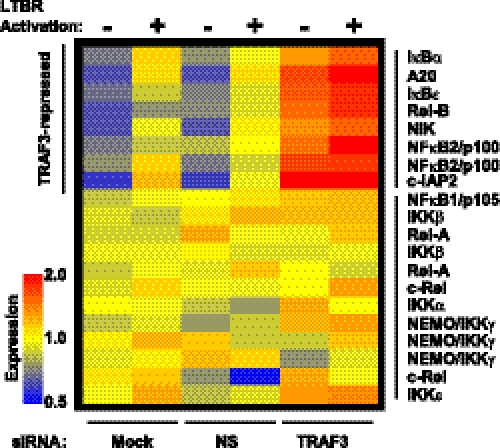
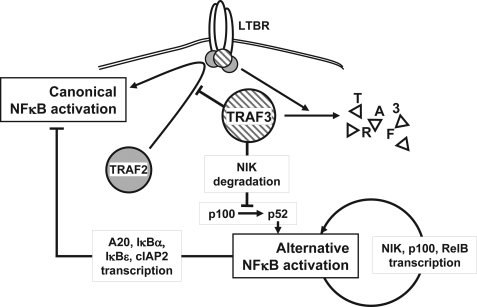
Components of lymphotoxin beta receptor (LTBR)-associated signaling complexes, including TRAF2, TRAF3, NIK, IKK1, and IKK2 have been shown to participate in the coupling of LTBR to NFkappaB. Here, we report that TRAF3 functions as a negative regulator of LTBR signaling via both canonical and non-canonical NFkappaB pathways by two distinct mechanisms. Analysis of NFkappaB signaling in cell lines with functionally intact NFkappaB pathway but lacking LTBR-mediated induction of NFkappaB target genes revealed an inverse association of cellular TRAF3 levels with LTBR-specific defect in canonical NFkappaB activation. Increased expression of TRAF3 correlated with its increased recruitment to LTBR-induced signaling complexes, decreased recruitment of TRAF2, and attenuated phosphorylation of IkappaB alpha and RelA. In contrast, activation of NFkappaB by TNF did not depend on TRAF3 levels. siRNA-mediated depletion of TRAF3 promoted recruitment of TRAF2 and IKK1 to activated LTBR, enabling LTBR-inducible canonical NFkappaB signaling and NFkappaB target gene expression. TRAF3 knock-down also increased mRNA and protein expression of several non-canonical NFkappaB components, including NFkappaB2/p100, RelB, and NIK, accompanied by processing of NFkappaB2/p100 into p52. These effects of TRAF3 depletion did not require LTBR signaling and were consistent with autonomous activation of the non-canonical NFkappaB pathway. Our data illustrate the function of TRAF3 as a dual-mode repressor of LTBR signaling that controls activation of canonical NFkappaB, and de-repression of the intrinsic activity of non-canonical NFkappaB. Modulation of cellular TRAF3 levels may thus contribute to regulation of NFkappaB-dependent gene expression by LTBR by affecting the balance of LTBR-dependent activation of canonical and non-canonical NFkappaB pathways.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
