

Atomic interaction networks in the core of protein domains and their native folds
- PMID: 20186337
- PMCID: PMC2826414
- DOI: 10.1371/journal.pone.0009391
Atomic interaction networks in the core of protein domains and their native folds
Abstract
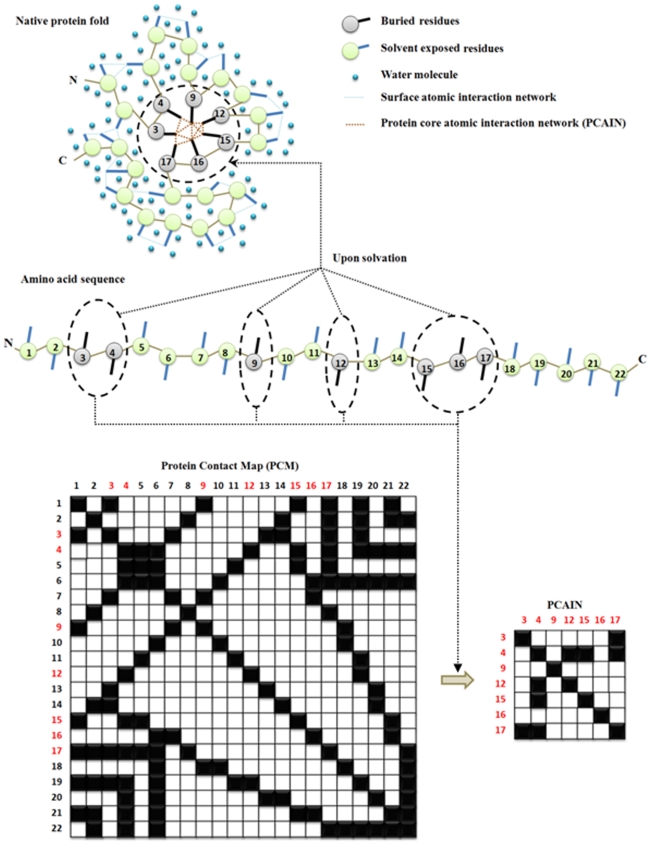
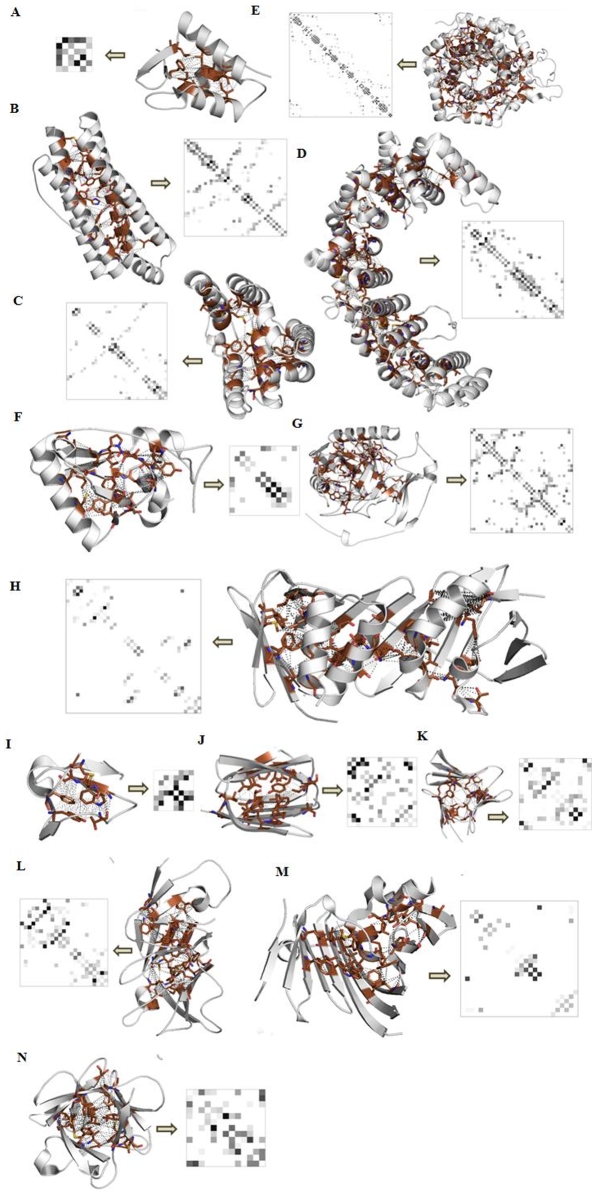
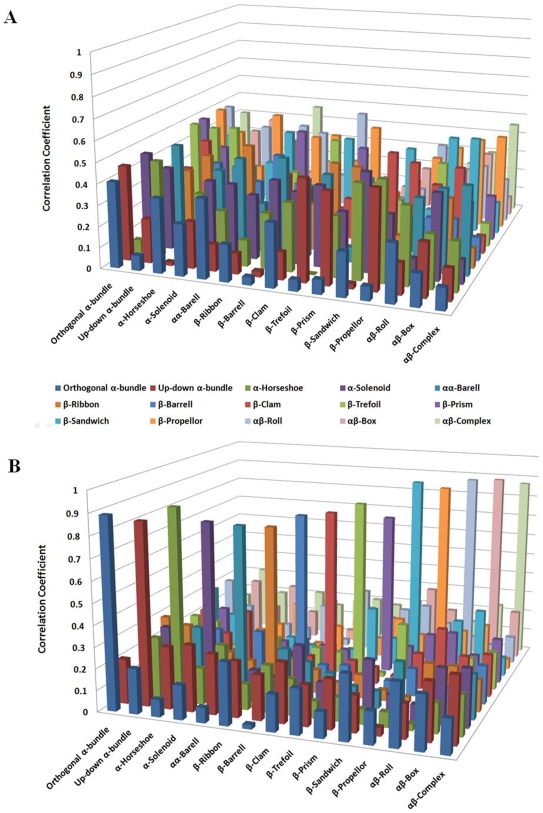
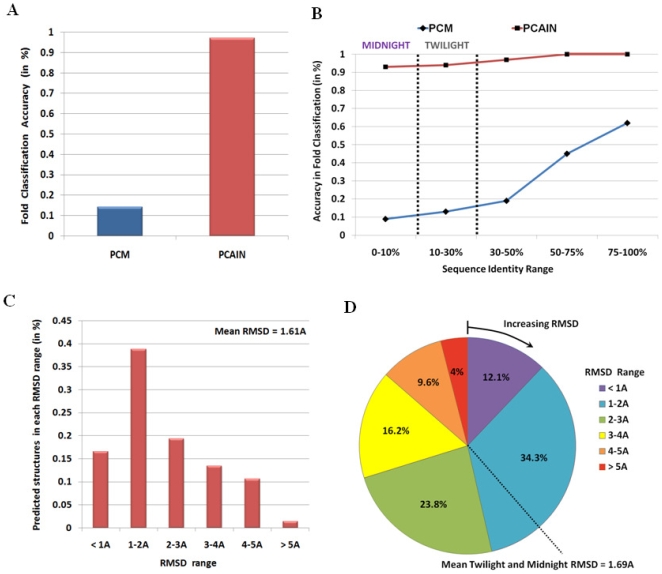
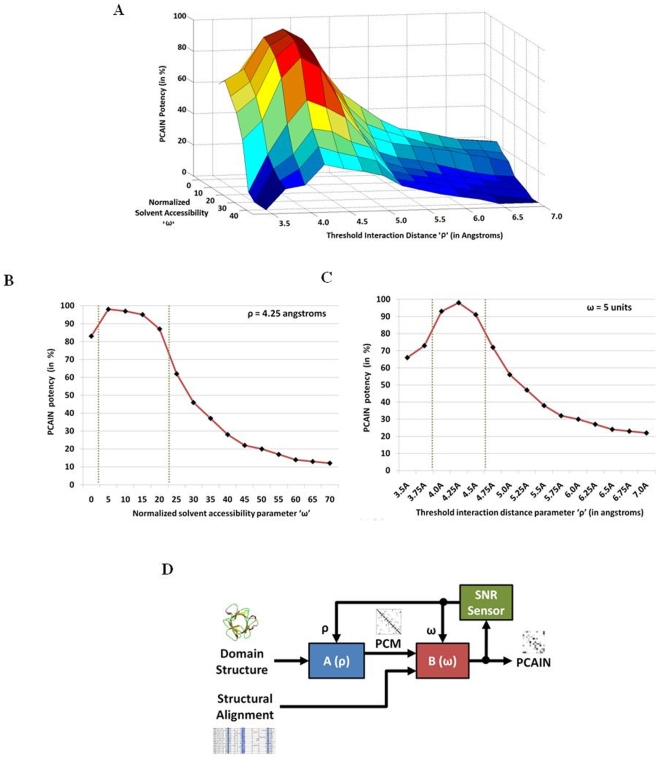
Vastly divergent sequences populate a majority of protein folds. In the quest to identify features that are conserved within protein domains belonging to the same fold, we set out to examine the entire protein universe on a fold-by-fold basis. We report that the atomic interaction network in the solvent-unexposed core of protein domains are fold-conserved, extraordinary sequence divergence notwithstanding. Further, we find that this feature, termed protein core atomic interaction network (or PCAIN) is significantly distinguishable across different folds, thus appearing to be "signature" of a domain's native fold. As part of this study, we computed the PCAINs for 8698 representative protein domains from families across the 1018 known protein folds to construct our seed database and an automated framework was developed for PCAIN-based characterization of the protein fold universe. A test set of randomly selected domains that are not in the seed database was classified with over 97% accuracy, independent of sequence divergence. As an application of this novel fold signature, a PCAIN-based scoring scheme was developed for comparative (homology-based) structure prediction, with 1-2 angstroms (mean 1.61A) C(alpha) RMSD generally observed between computed structures and reference crystal structures. Our results are consistent across the full spectrum of test domains including those from recent CASP experiments and most notably in the 'twilight' and 'midnight' zones wherein <30% and <10% target-template sequence identity prevails (mean twilight RMSD of 1.69A). We further demonstrate the utility of the PCAIN protocol to derive biological insight into protein structure-function relationships, by modeling the structure of the YopM effector novel E3 ligase (NEL) domain from plague-causative bacterium Yersinia Pestis and discussing its implications for host adaptive and innate immune modulation by the pathogen. Considering the several high-throughput, sequence-identity-independent applications demonstrated in this work, we suggest that the PCAIN is a fundamental fold feature that could be a valuable addition to the arsenal of protein modeling and analysis tools.
Conflict of interest statement
Figures
Similar articles
-
The many faces of the YopM effector from plague causative bacterium Yersinia pestis and its implications for host immune modulation.Innate Immun. 2011 Dec;17(6):548-57. doi: 10.1177/1753425910377099. Epub 2010 Aug 10. Innate Immun. 2011. PMID: 20699282
-
CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures.PLoS Comput Biol. 2007 Nov;3(11):e232. doi: 10.1371/journal.pcbi.0030232. PLoS Comput Biol. 2007. PMID: 18052539 Free PMC article.
-
Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models.Proteins. 2005;61 Suppl 7:84-90. doi: 10.1002/prot.20723. Proteins. 2005. PMID: 16187348
-
IgStrand: A universal residue numbering scheme for the immunoglobulin-fold (Ig-fold) to study Ig-proteomes and Ig-interactomes.PLoS Comput Biol. 2025 Apr 14;21(4):e1012813. doi: 10.1371/journal.pcbi.1012813. eCollection 2025 Apr. PLoS Comput Biol. 2025. PMID: 40228037 Free PMC article. Review.
-
A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction.Curr Opin Struct Biol. 2005 Jun;15(3):285-9. doi: 10.1016/j.sbi.2005.05.011. Curr Opin Struct Biol. 2005. PMID: 15939584 Review.
Cited by
-
Predicting Designability of Small Proteins from Graph Features of Contact Maps.J Comput Biol. 2016 May;23(5):400-11. doi: 10.1089/cmb.2015.0209. J Comput Biol. 2016. PMID: 27159634 Free PMC article.
-
Contribution to the prediction of the fold code: application to immunoglobulin and flavodoxin cases.PLoS One. 2015 Apr 27;10(4):e0125098. doi: 10.1371/journal.pone.0125098. eCollection 2015. PLoS One. 2015. PMID: 25915049 Free PMC article.
-
Cutoff Scanning Matrix (CSM): structural classification and function prediction by protein inter-residue distance patterns.BMC Genomics. 2011 Dec 22;12 Suppl 4(Suppl 4):S12. doi: 10.1186/1471-2164-12-S4-S12. Epub 2011 Dec 22. BMC Genomics. 2011. PMID: 22369665 Free PMC article.
-
Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment.Source Code Biol Med. 2015 May 22;10:7. doi: 10.1186/s13029-015-0037-3. eCollection 2015. Source Code Biol Med. 2015. PMID: 26089981 Free PMC article.
-
Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas.Nat Struct Mol Biol. 2018 Feb;25(2):185-194. doi: 10.1038/s41594-017-0019-z. Epub 2018 Jan 15. Nat Struct Mol Biol. 2018. PMID: 29335563 Free PMC article.
References
-
- Bloom JD, Drummond DA, Arnold FH, Wilke CO. Structural Determinants of the Rate of Protein Evolution in Yeast. Molecular Biology and Evolution. 2006;23(9):1751–1761. - PubMed
-
- Pratt LR, Chandler D. Theory of the hydrophobic effect. J Chem Phys. 1977;67:3683–3704.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
