

22q13.3 deletion syndrome: clinical and molecular analysis using array CGH
- PMID: 20186804
- PMCID: PMC3119894
- DOI: 10.1002/ajmg.a.33253
22q13.3 deletion syndrome: clinical and molecular analysis using array CGH
Abstract
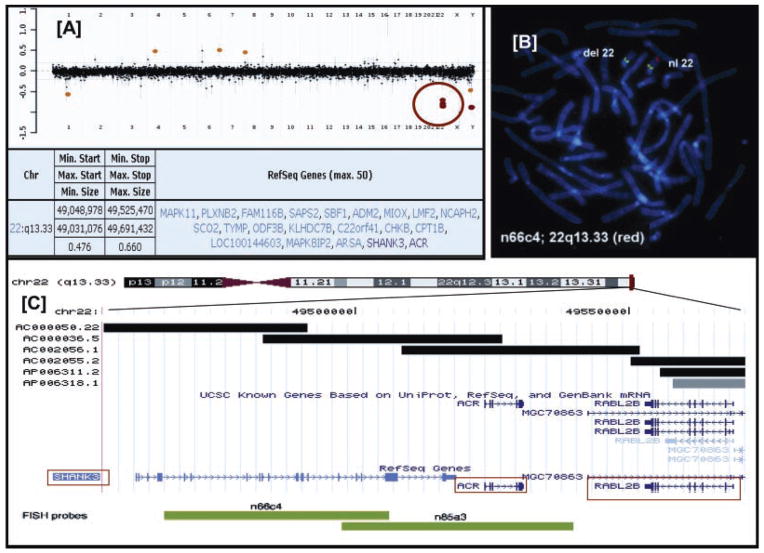
The 22q13.3 deletion syndrome results from loss of terminal segments of varying sizes at 22qter. Few genotype-phenotype correlations have been found but all patients have mental retardation and severe delay, or absence of, expressive speech. We carried out clinical and molecular characterization of 13 patients. Developmental delay and speech abnormalities were common to all and comparable in frequency and severity to previously reported cases. Array-based comparative genomic hybridization showed the deletions to vary from 95 kb to 8.5 Mb. We also carried out high-resolution 244K array comparative genomic hybridization in 10 of 13 patients, that defined the proximal and distal breakpoints of each deletion and helped determine the size, extent, and gene content within the deletion. Two patients had a smaller 95 kb terminal deletion with breakpoints within the SHANK3 gene while three other patients had a similar 5.5 Mb deletion implying the recurrent nature of these deletions. The two largest deletions were found in patients with ring chromosome 22. No correlation could be made with deletion size and phenotype although complete/partial SHANK3 was deleted in all patients. There are very few reports on array comparative genomic hybridization analysis on patients with the 22q13.3 deletion syndrome, and we aim to accurately characterize these patients both clinically and at the molecular level, to pave the way for further genotype-phenotype correlations. (c) 2010 Wiley-Liss, Inc.
Figures


References
-
- Anderlid BM, Schoumans J, Anneren G, Tapia-Paez I, Dumanski J, Blennow E, Nordenskjold M. FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet. 2002;110:439–443. - PubMed
-
- Ching TT, Maunakea AK, Jun P, Hong C, Zardo G, Pinkel D, Albertson DG, Fridlyand J, Mao J-H, Shchors K, Weiss WA, Costello JF. Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3. Nat Genet. 2005;37:645–651. - PubMed
-
- Cusmano-Ozog K, Manning MA, Hoyme HE. 22q13.3 deletion syndrome: A recognizable malformation syndrome associated with marked speech and language delay. Am J Med Genet Part C. 2007;145C:393–398. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
