

Modeling expression quantitative trait loci in data combining ethnic populations
- PMID: 20187971
- PMCID: PMC2844390
- DOI: 10.1186/1471-2105-11-111
Modeling expression quantitative trait loci in data combining ethnic populations
Abstract
Background: Combining data from different ethnic populations in a study can increase efficacy of methods designed to identify expression quantitative trait loci (eQTL) compared to analyzing each population independently. In such studies, however, the genetic diversity of minor allele frequencies among populations has rarely been taken into account. Due to the fact that allele frequency diversity and population-level expression differences are present in populations, a consensus regarding the optimal statistical approach for analysis of eQTL in data combining different populations remains inconclusive.
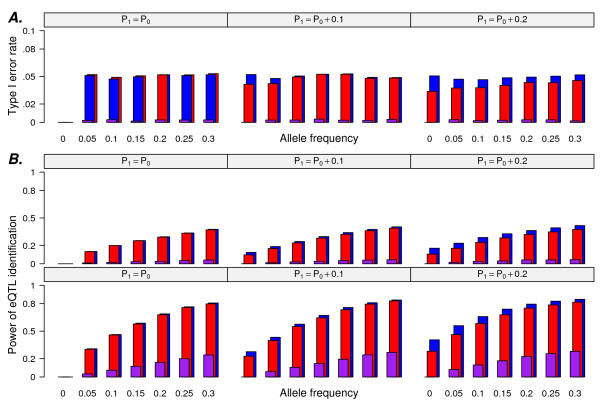
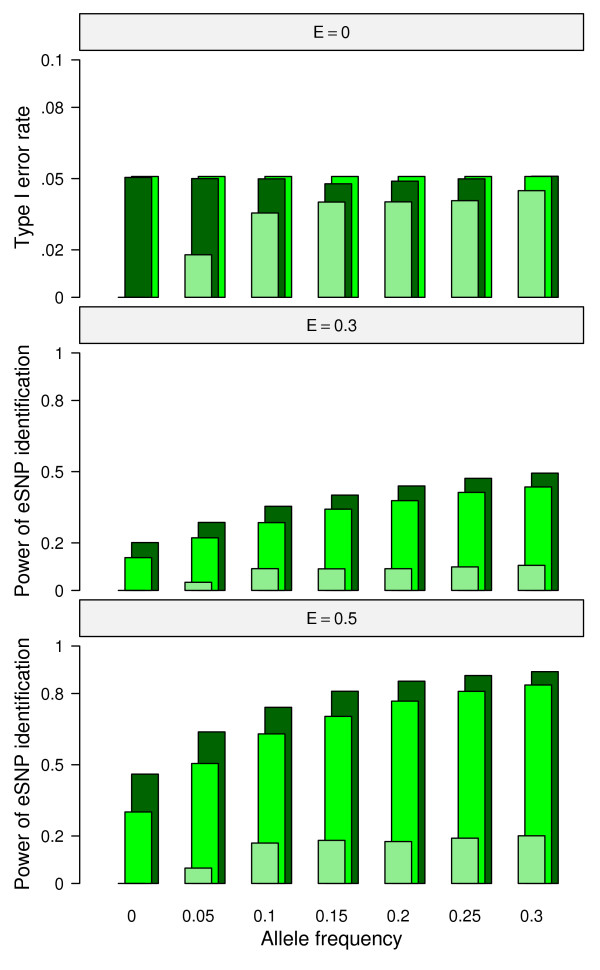
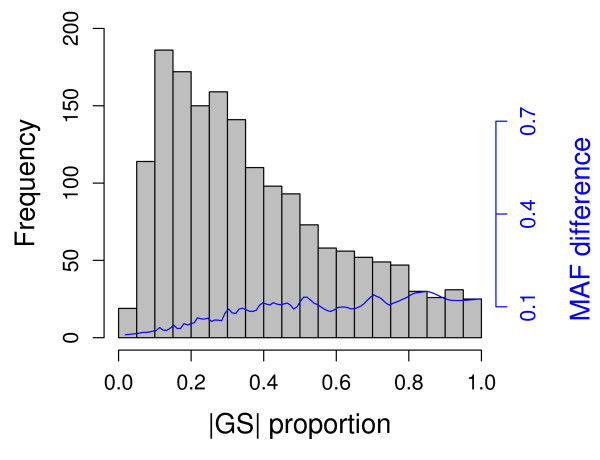
Results: In this report, we explored the applicability of a constrained two-way model to identify eQTL for combined ethnic data that might contain genetic diversity among ethnic populations. In addition, gene expression differences resulted from ethnic allele frequency diversity between populations were directly estimated and analyzed by the constrained two-way model. Through simulation, we investigated effects of genetic diversity on eQTL identification by examining gene expression data pooled from normal quantile transformation of each population. Using the constrained two-way model to reanalyze data from Caucasians and Asian individuals available from HapMap, a large number of eQTL were identified with similar genetic effects on the gene expression levels in these two populations. Furthermore, 19 single nucleotide polymorphisms with inter-population differences with respect to both genotype frequency and gene expression levels directed by genotypes were identified and reflected a clear distinction between Caucasians and Asian individuals.
Conclusions: This study illustrates the influence of minor allele frequencies on common eQTL identification using either separate or combined population data. Our findings are important for future eQTL studies in which different datasets are combined to increase the power of eQTL identification.
Figures
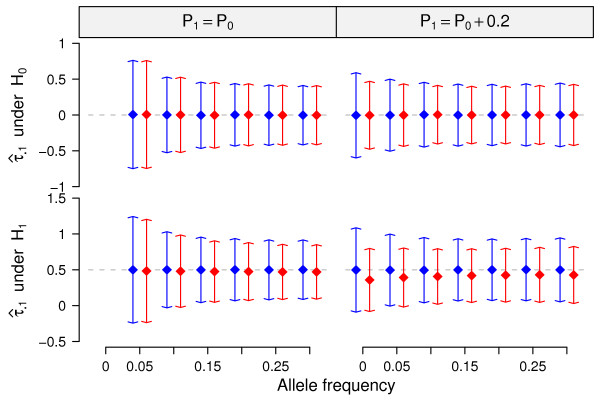
 estimated by CTWM (blue) and QT (red) methods under null (upper panel, E = 0) and alternative (lower panel, E = 0.5) hypotheses. Arrows of each dot represent the 95% confidence interval calculated from 10,000 simulations. Dash lines are the true values of τ•1 used in the simulations.
estimated by CTWM (blue) and QT (red) methods under null (upper panel, E = 0) and alternative (lower panel, E = 0.5) hypotheses. Arrows of each dot represent the 95% confidence interval calculated from 10,000 simulations. Dash lines are the true values of τ•1 used in the simulations.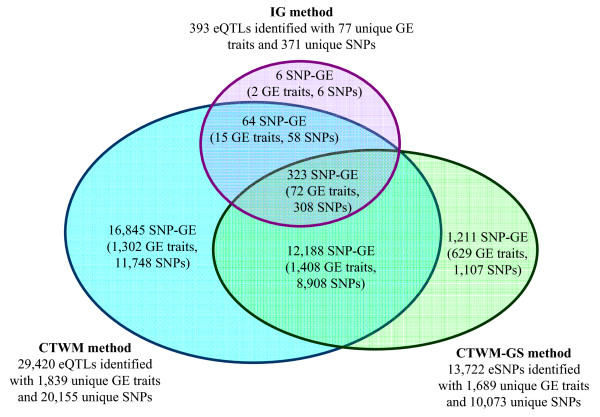

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
