

Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta
- PMID: 20188343
- PMCID: PMC2833387
- DOI: 10.1016/j.ajhg.2010.01.034
Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta
Abstract
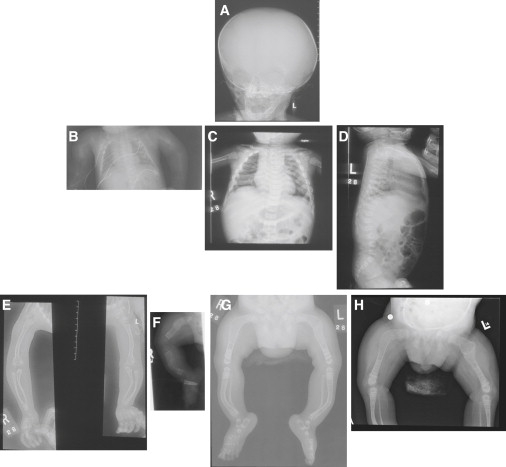
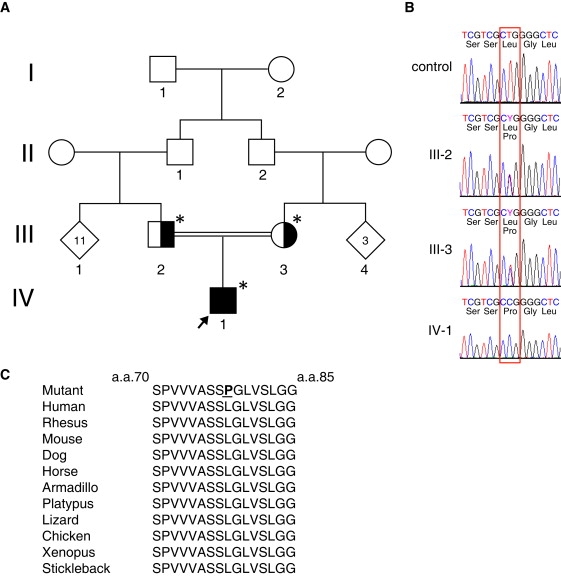
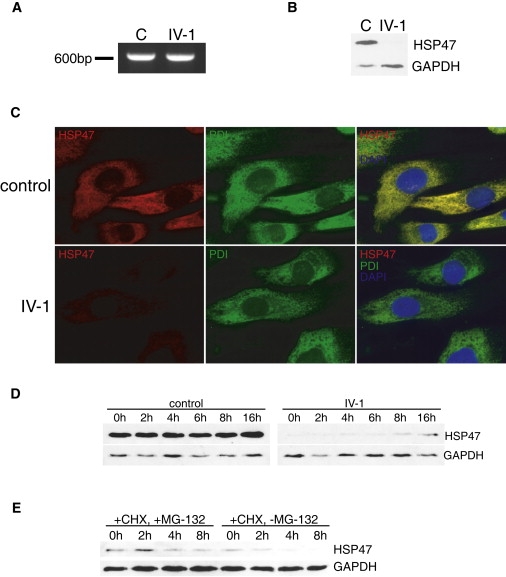
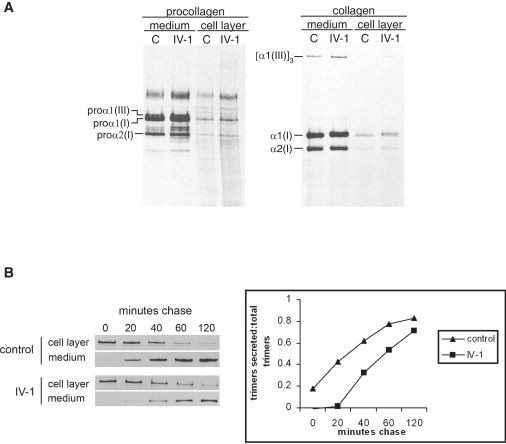
Osteogenesis imperfecta (OI) is characterized by bone fragility and fractures that may be accompanied by bone deformity, dentinogenesis imperfecta, short stature, and shortened life span. About 90% of individuals with OI have dominant mutations in the type I collagen genes COL1A1 and COL1A2. Recessive forms of OI resulting from mutations in collagen-modifying enzymes and chaperones CRTAP, LEPRE1, PPIB, and FKBP10 have recently been identified. We have identified an autosomal-recessive missense mutation (c.233T>C, p.Leu78Pro) in SERPINH1, which encodes the collagen chaperone-like protein HSP47, that leads to a severe OI phenotype. The mutation results in degradation of the endoplasmic reticulum resident HSP47 via the proteasome. Type I procollagen accumulates in the Golgi of fibroblasts from the affected individual and a population of the secreted type I procollagen is protease sensitive. These findings suggest that HSP47 monitors the integrity of the triple helix of type I procollagen at the ER/cis-Golgi boundary and, when absent, the rate of transit from the ER to the Golgi is increased and helical structure is compromised. The normal 3-hydroxylation of the prolyl residue at position 986 of the triple helical domain of proalpha1(I) chains places the role of HSP47 downstream from the CRTAP/P3H1/CyPB complex that is involved in prolyl 3-hydroxylation. Identification of this mutation in SERPINH1 gives further insight into critical steps of the collagen biosynthetic pathway and the molecular pathogenesis of OI.
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Byers P.H., Cole W.G. Osteogenesis imperfecta. In: Royce P.M., Steinmann B., editors. Connective Tissue and It's Heritable Disorders: Molecular, Genetic, and Medical Aspects. Wiley-Liss, Inc.; New York: 2002. pp. 385–430.
-
- Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. - PubMed
-
- Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S., Hyland J.C., Körkkö J., Prockop D.J., De Paepe A. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. - PMC - PubMed
-
- Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
