

Opioid pharmaceuticals and addiction: the issues, and research directions seeking solutions
- PMID: 20188495
- PMCID: PMC3072810
- DOI: 10.1016/j.drugalcdep.2010.01.001
Opioid pharmaceuticals and addiction: the issues, and research directions seeking solutions
Abstract
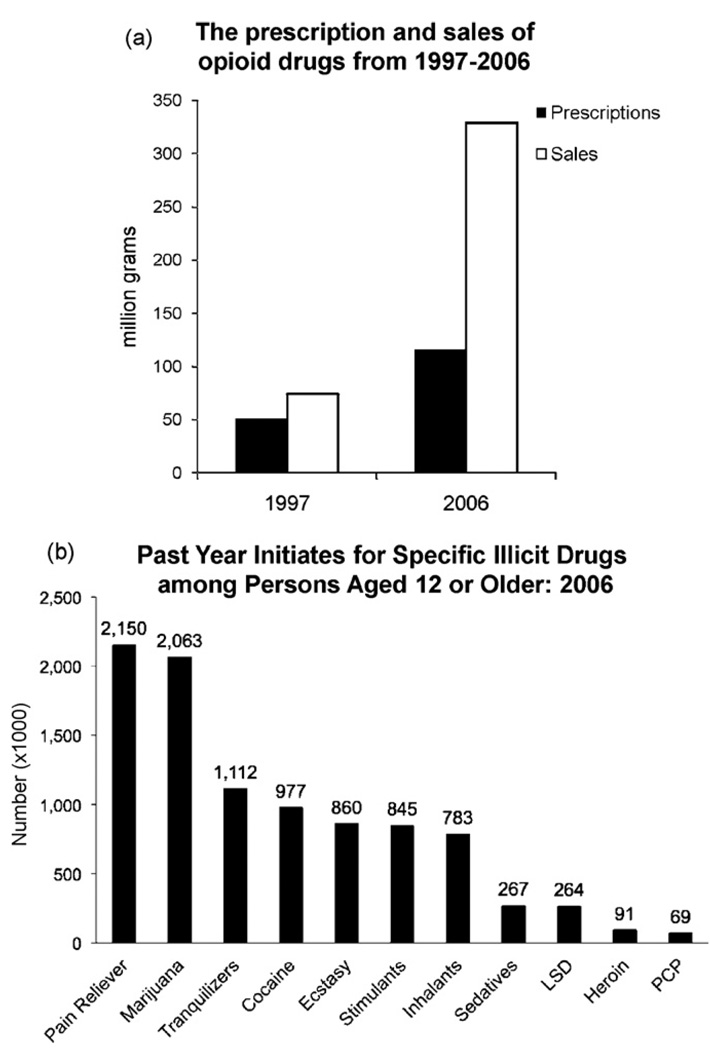
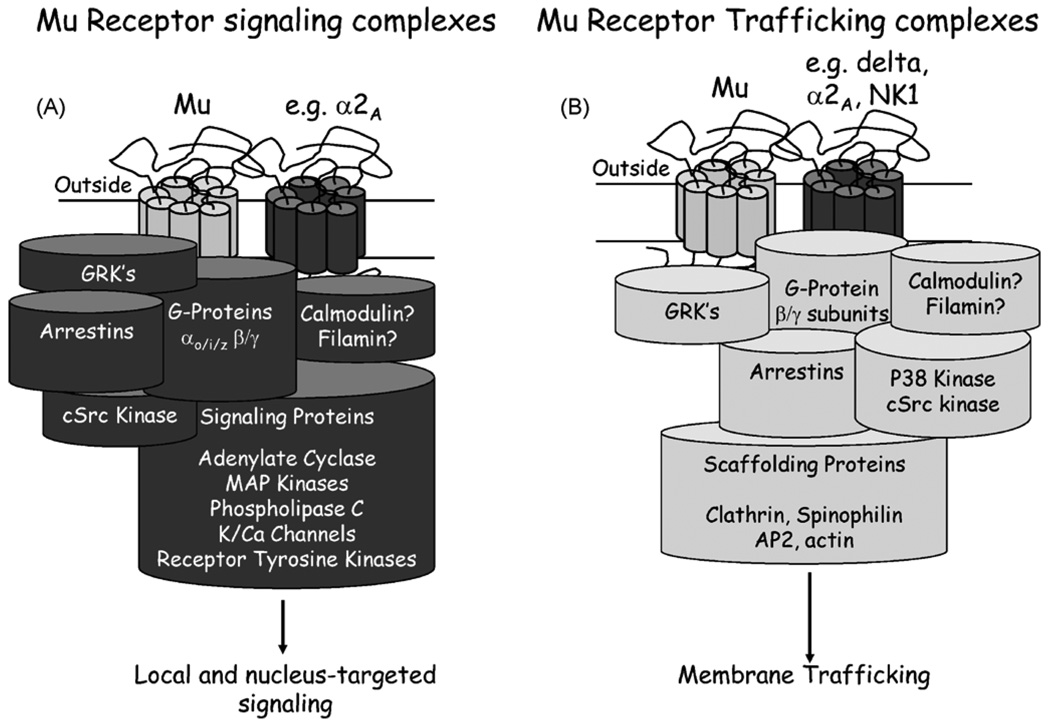
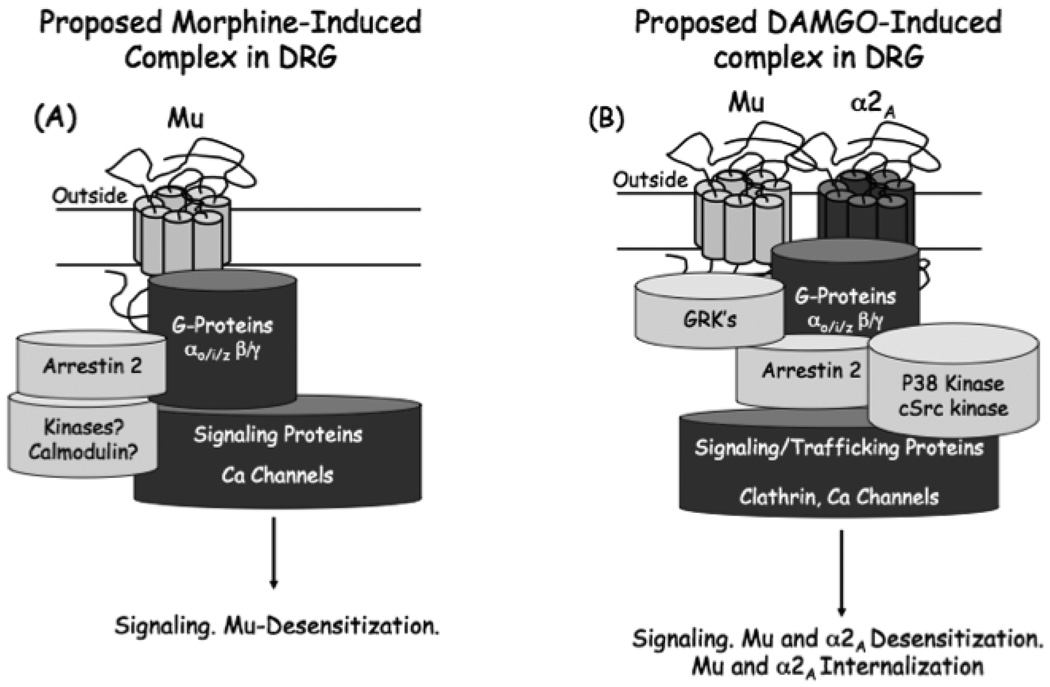
There are few pharmaceuticals superior to opiates for the treatment of pain. However, with concerns of addiction, withdrawal and questionable efficacy for all types of pain, these compounds are far from a magical panacea for pain-relief. As it is unlikely that other classes of compounds will supersede the opioids in the very near future, it is important to both optimize current opioid therapies and curb the astounding diversion of opioids from their intended analgesic use to non-medical abuse. In optimizing opioid therapeutics it is necessary to enhance the clinical awareness of the benefits of treating pain and combine this with aggressive strategies to reduce diversion for non-medical use. At the heart of the issue of opioid misuse is the role of opioid systems in the reward circuitry, and the adaptive processes associated with repetitive opioid use that manifest during withdrawal. Emerging pharmacological insights of opioid receptors will be reviewed that provide future hope for developing opioid-based analgesics with reduced addictive properties and perhaps, reduced opponent processes. In addition, with the increased understanding of nociceptive circuitry and the molecules involved in transmitting pain, new therapeutic targets have become evident that may result in effective analgesics either alone or in combination with current opioid therapies.
Conflict of interest statement
These authors have no conflict of interest.
Figures
Comment in
-
Overview and historical perspective of four papers presented on research related to the endogenous opioid system.Drug Alcohol Depend. 2010 May 1;108(3):195-9. doi: 10.1016/j.drugalcdep.2010.03.003. Epub 2010 Apr 18. Drug Alcohol Depend. 2010. PMID: 20399574 Free PMC article. No abstract available.
References
-
- Ashburn MA, Rubingh CR. The role of non-opioid analgesics for the management of postoperative pain. Cancer Control. 1999;6:10–13. - PubMed
-
- Ashburn MA, Staats PS. Management of chronic pain. Lancet. 1999;353:1865–1869. - PubMed
-
- Ballantyne JC, Mao J. Opioid therapy for chronic pain. N. Engl. J. Med. 2003;349:1943–1953. - PubMed
-
- Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71:559–566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
