

Immunodeficiency due to mutations in ORAI1 and STIM1
- PMID: 20189884
- PMCID: PMC2856745
- DOI: 10.1016/j.clim.2010.01.011
Immunodeficiency due to mutations in ORAI1 and STIM1
Abstract
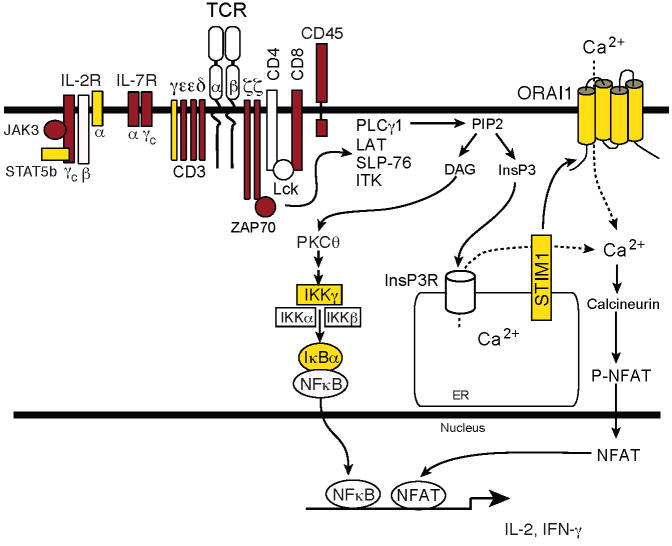
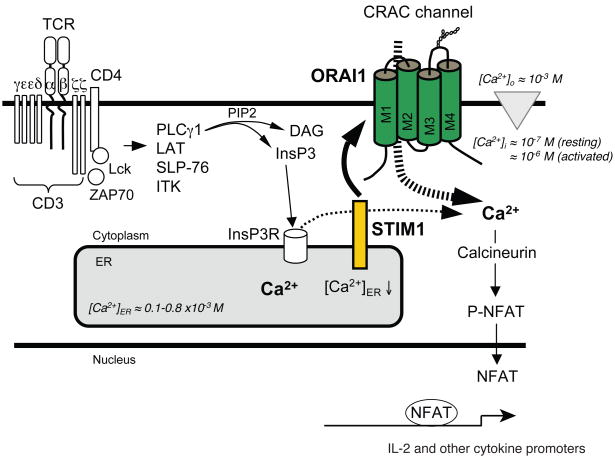
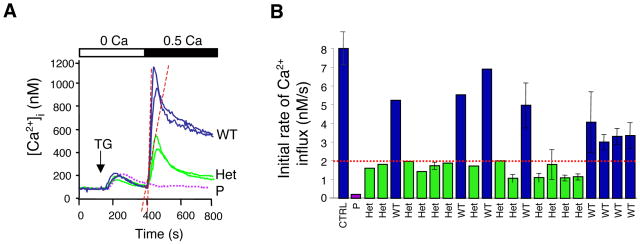
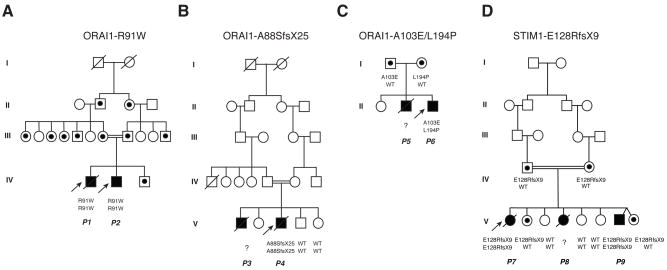
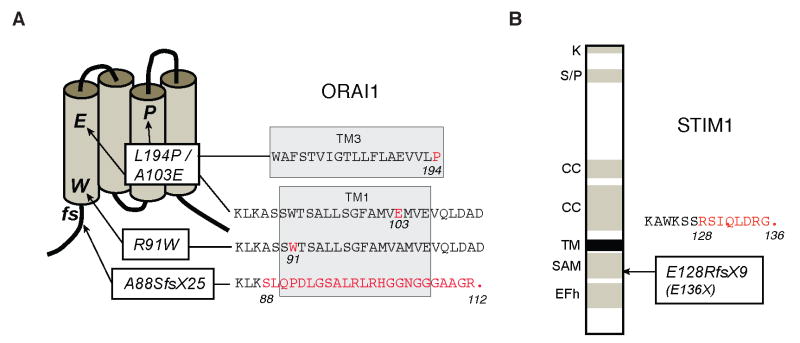
Lymphocyte activation requires Ca(2+) influx through specialized Ca(2+) channels in the plasma membrane. In T cells the predominant Ca(2+) channel is the Ca(2+) release activated Ca(2+) (CRAC) channel encoded by the gene ORAI1. ORAI1 is activated by stromal interaction molecule (STIM) 1 that is localized in the ER where it senses the concentration of stored Ca(2+). Following antigen binding to immunoreceptors such as the TCR, ER Ca(2+) stores are depleted, STIM1 is activated and ORAI1-CRAC channels open resulting in what is referred to as store-operated Ca(2+) entry (SOCE). Mutations in ORAI1 and STIM1 genes in human patients that lead to expression of non-functional ORAI1 or complete lack of ORAI1 or STIM1 protein are associated with a unique clinical phenotype that is characterized by immunodeficiency, muscular hypotonia and anhydrotic ectodermal dysplasia, as well as, in the case of STIM1 deficiency, autoimmunity and lymphoproliferative disease. The immunodeficiency in these patients is due to a severe defect in T cell activation but not in lymphocyte development. This review describes the immunological and non-immunological phenotypes of patients with defects in SOCE and CRAC channel function and discusses them in the context of similar immunodeficiency diseases and animal models of ORAI1 and STIM1 function.
Copyright 2008 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
