

Using a label-free proteomics method to identify differentially abundant proteins in closely related hypo- and hypervirulent clinical Mycobacterium tuberculosis Beijing isolates
- PMID: 20190197
- PMCID: PMC2984234
- DOI: 10.1074/mcp.M900422-MCP200
Using a label-free proteomics method to identify differentially abundant proteins in closely related hypo- and hypervirulent clinical Mycobacterium tuberculosis Beijing isolates
Abstract
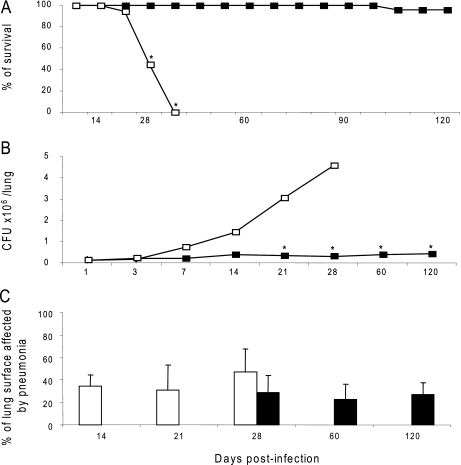
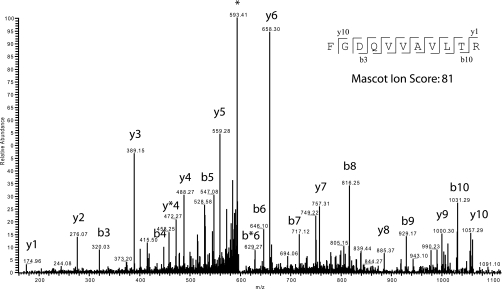
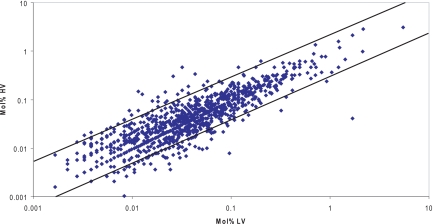
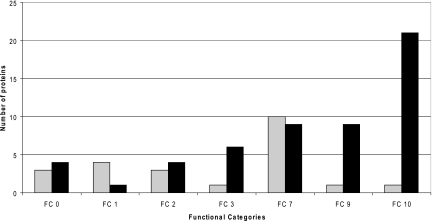
Although the genome of the Mycobacterium tuberculosis H37Rv laboratory strain has been available for over 10 years, it is only recently that genomic information from clinical isolates has been used to generate the hypothesis of virulence differences between different strains. In addition, the relationship between strains displaying differing virulence in an epidemiological setting and their behavior in animal models has received little attention. The potential causes for variation in virulence between strains, as determined by differential protein expression, have similarly been a neglected area of investigation. In this study, we used a label-free quantitative proteomics approach to estimate differences in protein abundance between two closely related Beijing genotypes that have been shown to be hyper- and hypovirulent on the basis of both epidemiological and mouse model studies. We were able to identify a total of 1668 proteins from both samples, and protein abundance calculations revealed that 48 proteins were over-represented in the hypovirulent isolate, whereas 53 were over-represented in the hypervirulent. Functional classification of these results shows that molecules of cell wall organization and DNA transcription regulatory proteins may have a critical influence in defining the level of virulence. The reduction in the presence of ESAT-6, other Esx-like proteins, and FbpD (MPT51) in the hypervirulent strain indicates that changes in the repertoire of highly immunogenic proteins can be a defensive process undertaken by the virulent cell. In addition, most of the previously well characterized gene targets related to virulence were found to be similarly expressed in our model. Our data support the use of proteomics as a complementary tool for genomic comparisons to understand the biology of M. tuberculosis virulence.
Figures

References
-
- Dormans J., Burger M., Aguilar D., Hernandez-Pando R., Kremer K., Roholl P., Arend S. M., van Soolingen D. (2004) Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin. Exp. Immunol. 137, 460–468 - PMC - PubMed
-
- Manca C., Tsenova L., Bergtold A., Freeman S., Tovey M., Musser J. M., Barry C. E., 3rd, Freedman V. H., Kaplan G. (2001) Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. U.S.A. 98, 5752–5757 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
