

Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization
- PMID: 20190753
- PMCID: PMC5308204
- DOI: 10.1038/ng.538
Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization
Erratum in
- Nat Genet. 2011 Mar;43(3):277
Abstract
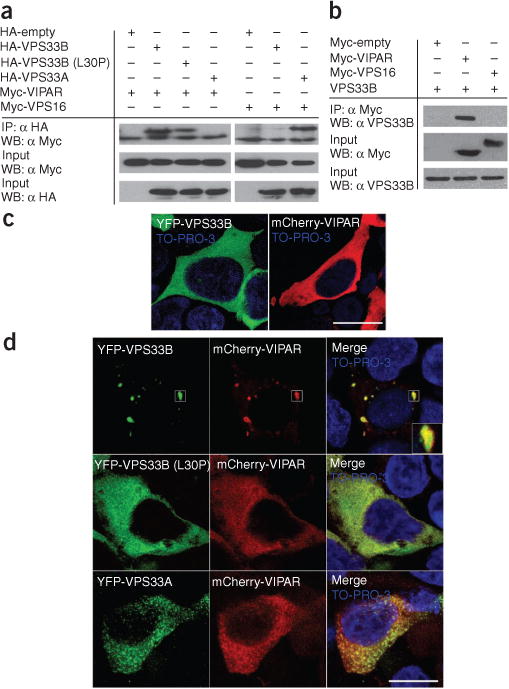
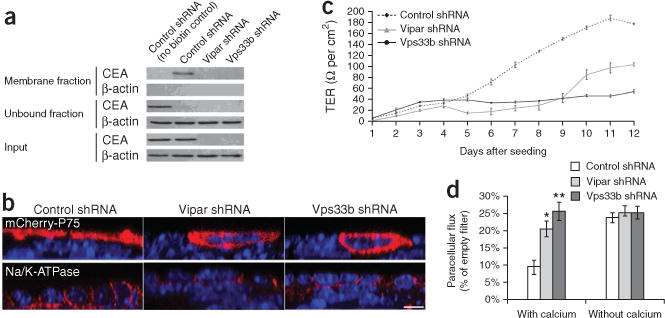
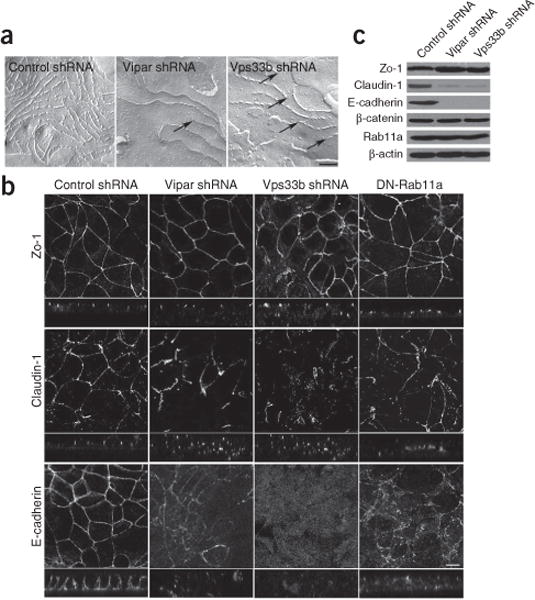
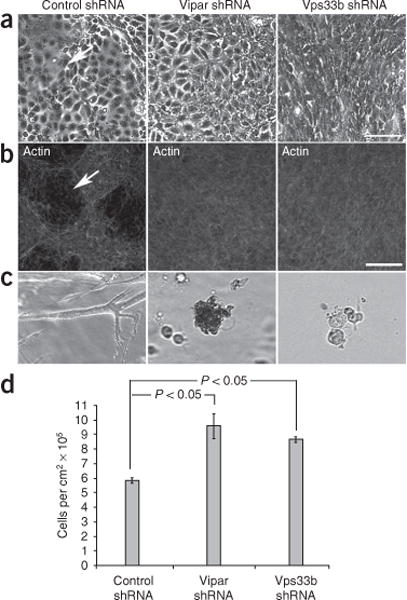
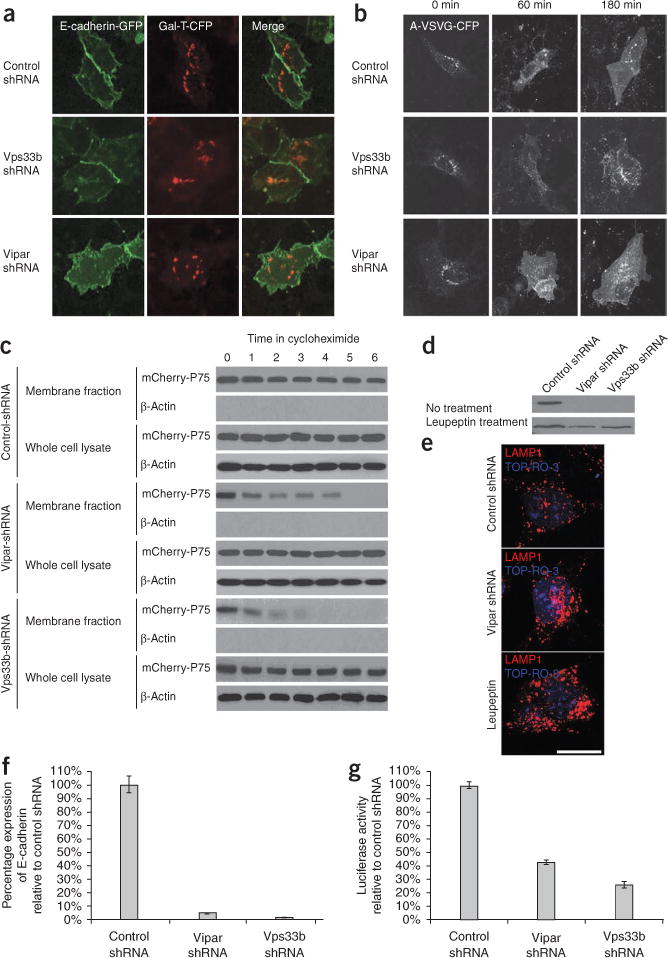
Arthrogryposis, renal dysfunction and cholestasis syndrome (ARC) is a multisystem disorder associated with abnormalities in polarized liver and kidney cells. Mutations in VPS33B account for most cases of ARC. We identified mutations in VIPAR (also called C14ORF133) in individuals with ARC without VPS33B defects. We show that VIPAR forms a functional complex with VPS33B that interacts with RAB11A. Knockdown of vipar in zebrafish resulted in biliary excretion and E-cadherin defects similar to those in individuals with ARC. Vipar- and Vps33b-deficient mouse inner medullary collecting duct (mIMDC-3) cells expressed membrane proteins abnormally and had structural and functional tight junction defects. Abnormal Ceacam5 expression was due to mis-sorting toward lysosomal degradation, but reduced E-cadherin levels were associated with transcriptional downregulation. The VPS33B-VIPAR complex thus has diverse functions in the pathways regulating apical-basolateral polarity in the liver and kidney.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


Comment in
-
On the endosomal function and gene nomenclature of human SPE-39.Nat Genet. 2011 Mar;43(3):176. doi: 10.1038/ng0311-176. Nat Genet. 2011. PMID: 21350494 No abstract available.
Similar articles
-
Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features.Ital J Pediatr. 2014 Sep 20;40:77. doi: 10.1186/s13052-014-0077-3. Ital J Pediatr. 2014. PMID: 25239142 Free PMC article. Review.
-
VIPAS39 related arthrogryposis-renal dysfunction-cholestasis syndrome-case report and systematic review.Orphanet J Rare Dis. 2024 Dec 30;19(1):496. doi: 10.1186/s13023-024-03486-2. Orphanet J Rare Dis. 2024. PMID: 39736737 Free PMC article.
-
Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome.Hum Mutat. 2012 Dec;33(12):1656-64. doi: 10.1002/humu.22155. Epub 2012 Aug 6. Hum Mutat. 2012. PMID: 22753090 Free PMC article.
-
VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function.Biochim Biophys Acta Mol Basis Dis. 2018 May;1864(5 Pt A):1609-1621. doi: 10.1016/j.bbadis.2018.01.028. Epub 2018 Jan 31. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 29409756 Free PMC article.
-
Zebrafish vps33b, an ortholog of the gene responsible for human arthrogryposis-renal dysfunction-cholestasis syndrome, regulates biliary development downstream of the onecut transcription factor hnf6.Development. 2005 Dec;132(23):5295-306. doi: 10.1242/dev.02140. Development. 2005. PMID: 16284120
Cited by
-
Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome: a rare association with high GGT level and absent kidney.BMJ Case Rep. 2018 Aug 9;2018:bcr2017223715. doi: 10.1136/bcr-2017-223715. BMJ Case Rep. 2018. PMID: 30093463 Free PMC article.
-
Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features.Ital J Pediatr. 2014 Sep 20;40:77. doi: 10.1186/s13052-014-0077-3. Ital J Pediatr. 2014. PMID: 25239142 Free PMC article. Review.
-
Making It New Again: Insight Into Liver Development, Regeneration, and Disease From Zebrafish Research.Curr Top Dev Biol. 2017;124:161-195. doi: 10.1016/bs.ctdb.2016.11.012. Epub 2017 Jan 17. Curr Top Dev Biol. 2017. PMID: 28335859 Free PMC article. Review.
-
Clathrin-dependent mechanisms modulate the subcellular distribution of class C Vps/HOPS tether subunits in polarized and nonpolarized cells.Mol Biol Cell. 2011 May 15;22(10):1699-715. doi: 10.1091/mbc.E10-10-0799. Epub 2011 Mar 16. Mol Biol Cell. 2011. PMID: 21411634 Free PMC article.
-
VIPAS39 related arthrogryposis-renal dysfunction-cholestasis syndrome-case report and systematic review.Orphanet J Rare Dis. 2024 Dec 30;19(1):496. doi: 10.1186/s13023-024-03486-2. Orphanet J Rare Dis. 2024. PMID: 39736737 Free PMC article.
References
-
- Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science. 1989;245:718–725. - PubMed
-
- Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. - PubMed
-
- Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. - PubMed
-
- Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol. 2004;16:140–145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
