

Interplay between Helicobacter pylori and immune cells in immune pathogenesis of gastric inflammation and mucosal pathology
- PMID: 20190789
- PMCID: PMC4003231
- DOI: 10.1038/cmi.2010.2
Interplay between Helicobacter pylori and immune cells in immune pathogenesis of gastric inflammation and mucosal pathology
Abstract
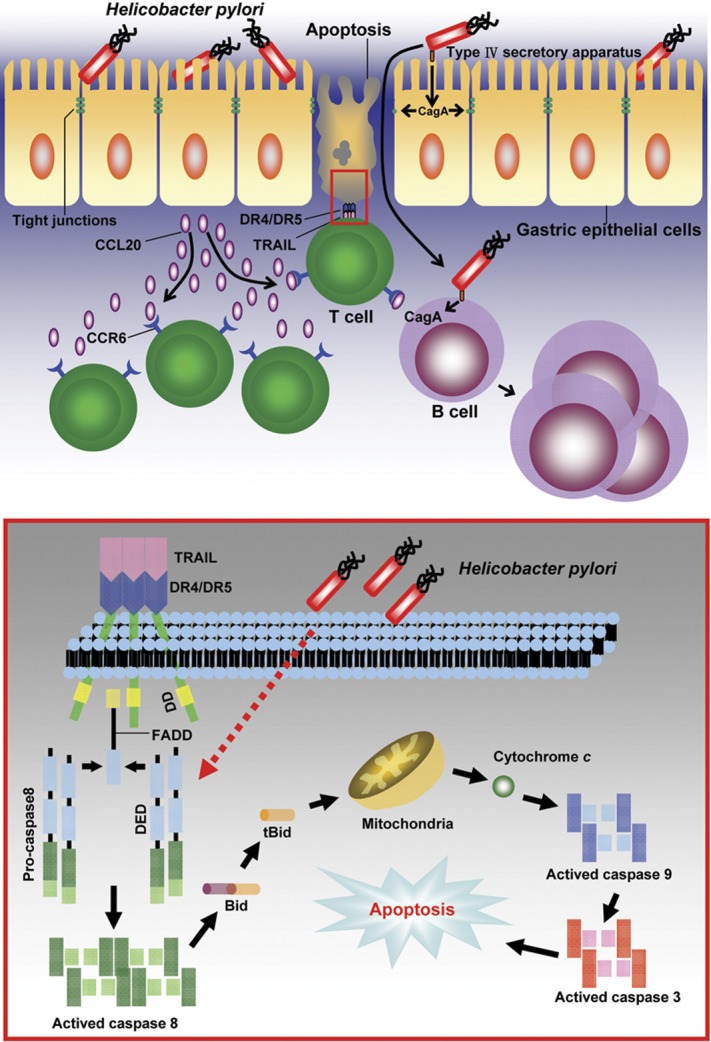
Helicobacter pylori infection is associated with an inflammatory response in the gastric mucosa, leading to chronic gastritis, peptic ulcers, gastric carcinoma and gastric mucosa-associated lymphoid tissue (MALT) lymphomas. Recent studies have shown that apoptosis of gastric epithelial cells is increased during H. pylori infection. Apoptosis induced by microbial infections are factors implicated in the pathogenesis of H. pylori infection. The enhanced gastric epithelial cell apoptosis in H. pylori infection has been suggested to play an important role in the pathogenesis of chronic gastritis and gastric pathology. In addition to directly triggering apoptosis, H. pylori induces sensitivity to tumor-necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in gastric epithelial cells via modulation of TRAIL apoptosis signaling. Moreover, H. pylori infection induces infiltration of T lymphocytes and triggers inflammation to augment apoptosis. In H. pylori infection, there was significantly increased CCR6(+)CD3(+ )T-cell infiltration in the gastric mucosa, and the CCR6 ligand, CCL20 chemokine, was selectively expressed in inflamed gastric tissues. These results implicate that the interaction between CCL20 and CCR6 may play a role in recruiting T cells to the sites of inflammation in the gastric mucosa during Helicobacter infection. Through these mechanisms, chemokine-mediated T lymphocyte trafficking into inflamed epithelium is initiated and the mucosal injury in Helicobacter infection is induced. This article will review the recent novel findings on the interactions of H. pylori with diverse host epithelial signaling pathways and events involved in the initiation of gastric pathology, including gastric inflammation, mucosal damage and development of MALT lymphomas.
Figures
References
-
- Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and risk of gastric carcinoma. N Engl J Med. 1994;325:1127–1131. - PubMed
-
- Parsonnet J. Molecular mechanisms for inflammation-promoted pathogenesis of cancer – The Sixteenth International Symposium of the Sapporo Cancer Seminar. Cancer Res. 1997;57:3620–3624. - PubMed
-
- Mannick EE, Bravo LE, Zarama G, Realpe JL, Zhang XJ, Ruiz B, et al. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res. 1996;56:3238–3243. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
