

A genomic approach to human autoimmune diseases
- PMID: 20192809
- PMCID: PMC2847838
- DOI: 10.1146/annurev-immunol-030409-101221
A genomic approach to human autoimmune diseases
Abstract
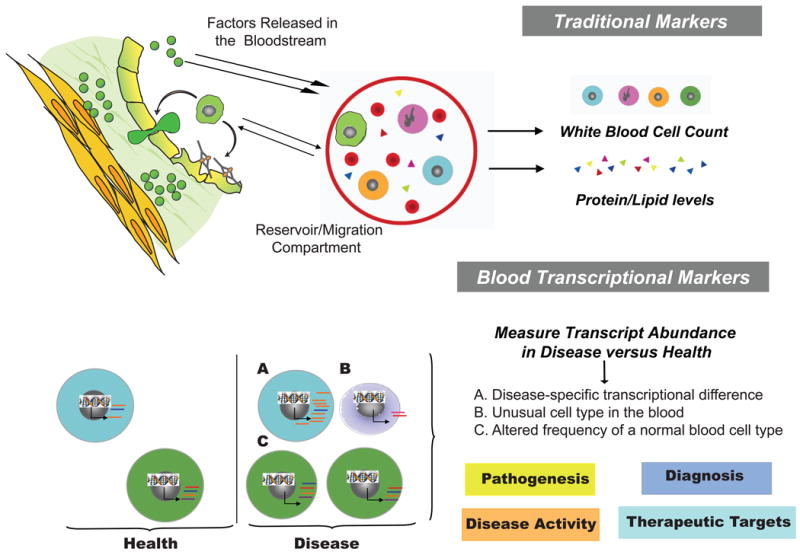
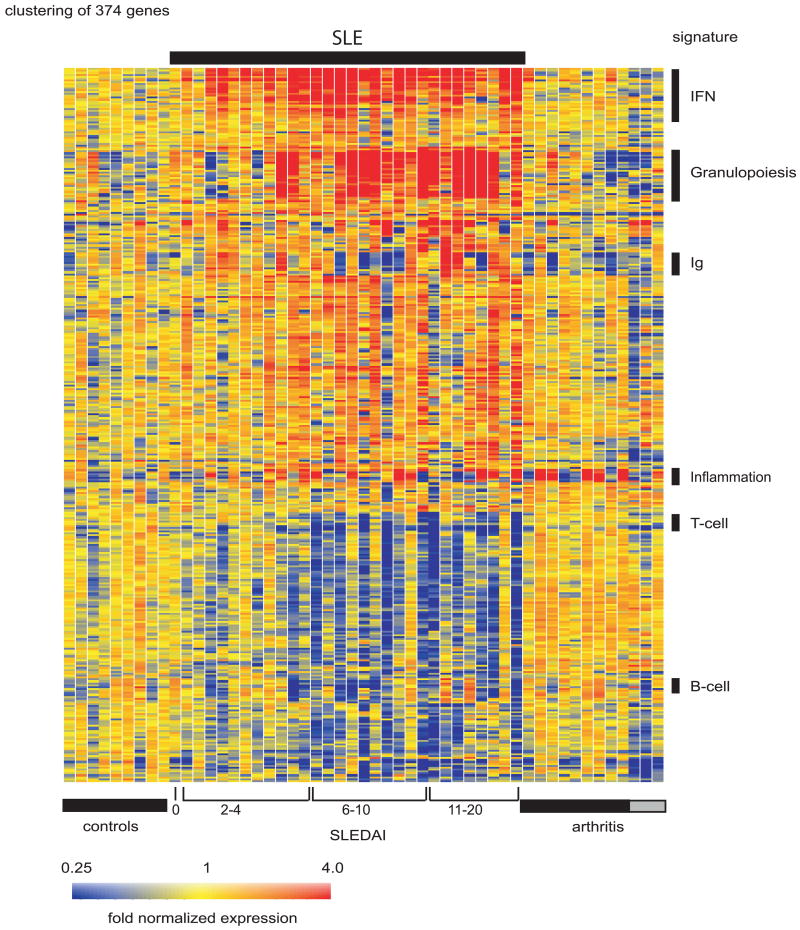
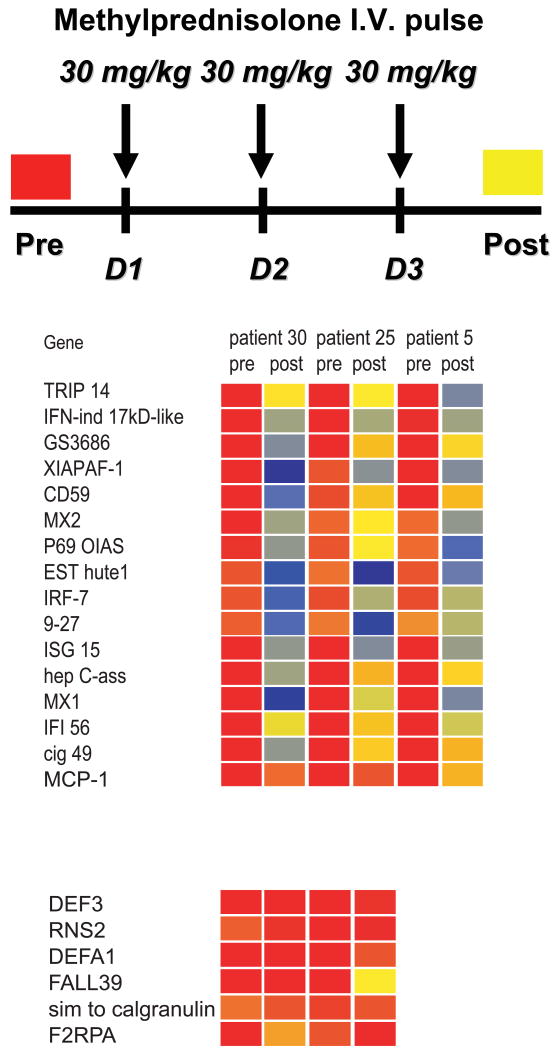
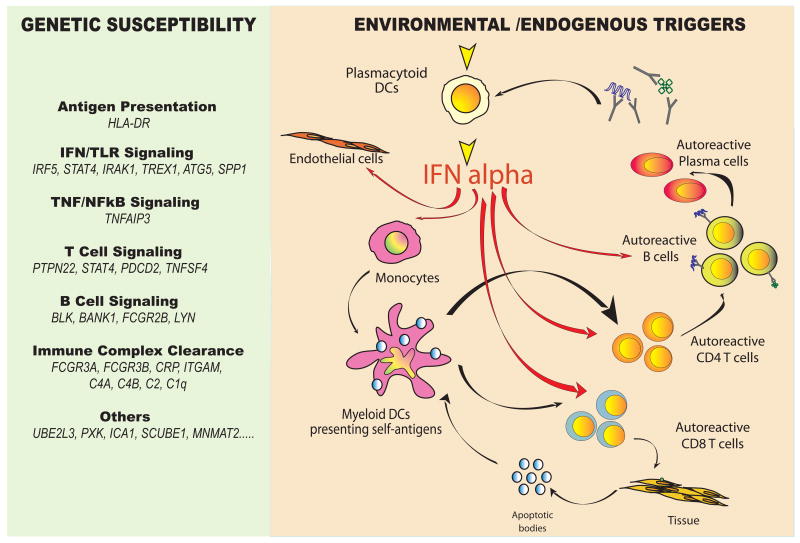
The past decade has seen an explosion in the use of DNA-based microarrays. These techniques permit assessment of RNA abundance on a genome-wide scale. Medical applications emerged in the field of cancer, with studies of both solid tumors and hematological malignancies leading to the development of tests that are now used to personalize therapeutic options. Microarrays have also been used to analyze the blood transcriptome in a wide range of diseases. In human autoimmune diseases, these studies are showing potential for identifying therapeutic targets as well as biomarkers for diagnosis, assessment of disease activity, and response to treatment. More quantitative and sensitive high-throughput RNA profiling methods are starting to be available and will be necessary for transcriptome analyses to become routine tests in the clinical setting. We expect this to crystallize within the coming decade, as these methods become part of the personalized medicine armamentarium.
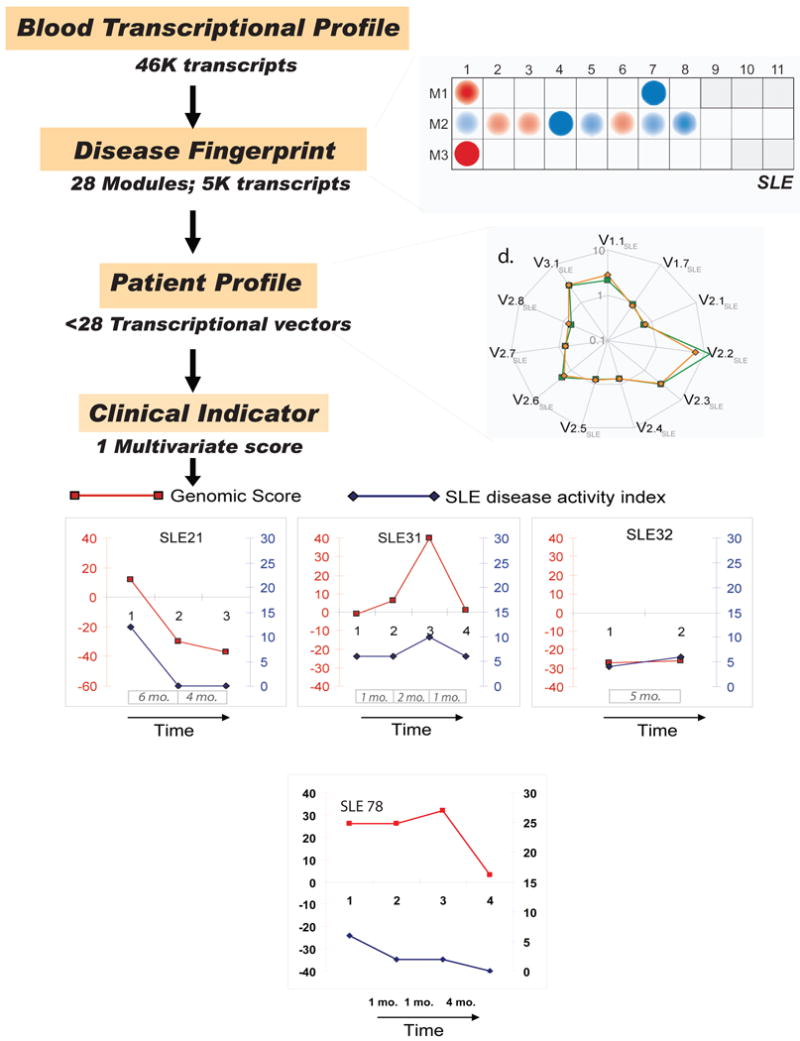
Figures
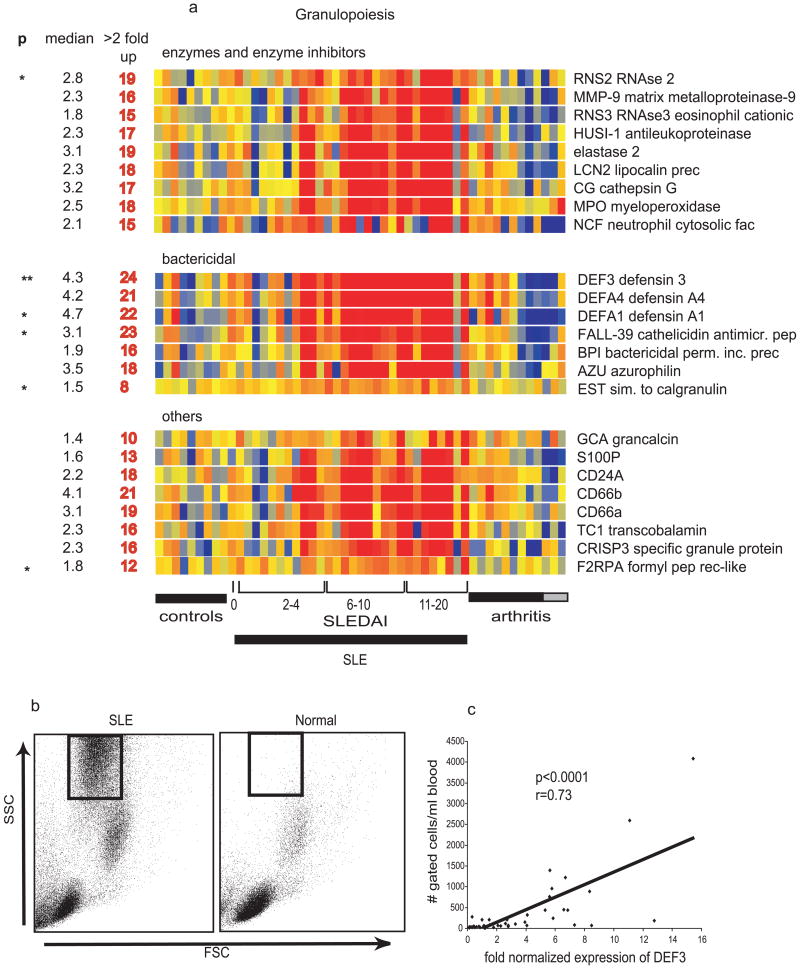
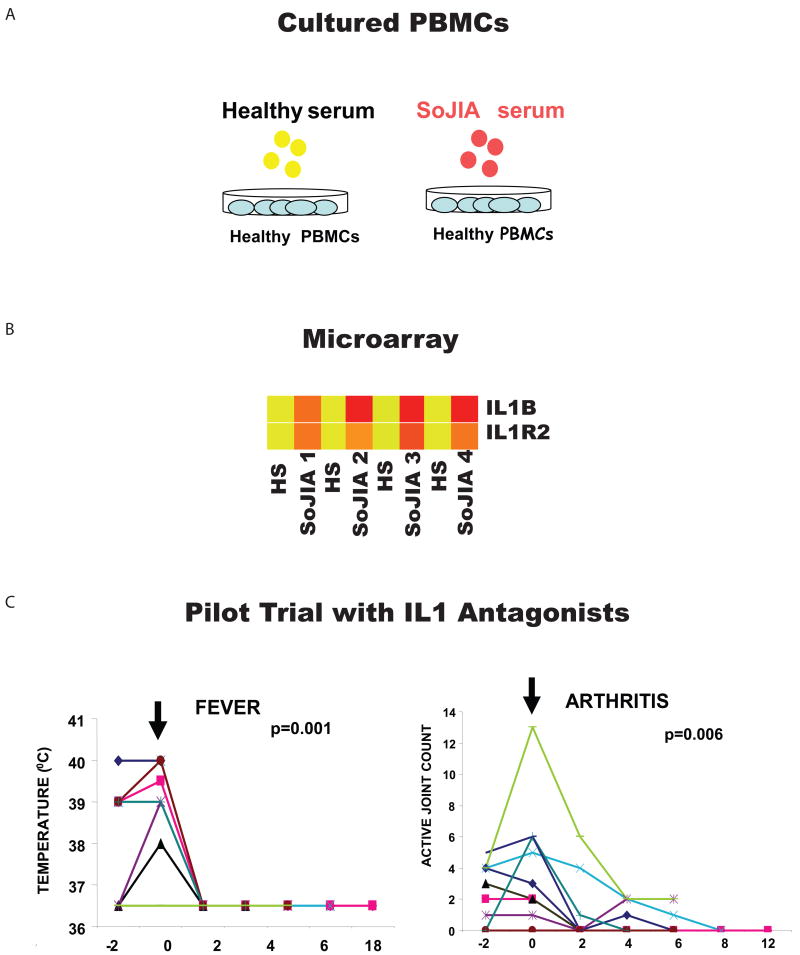
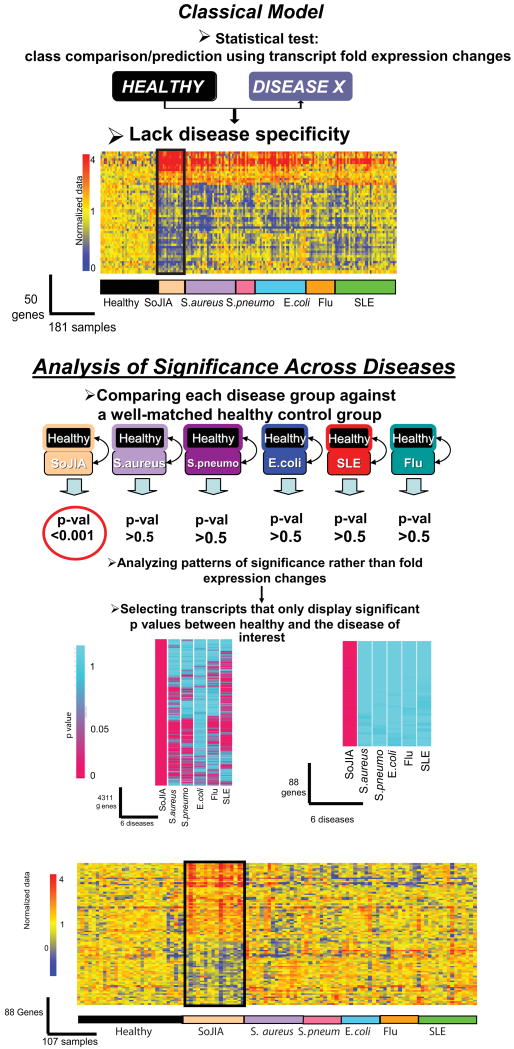
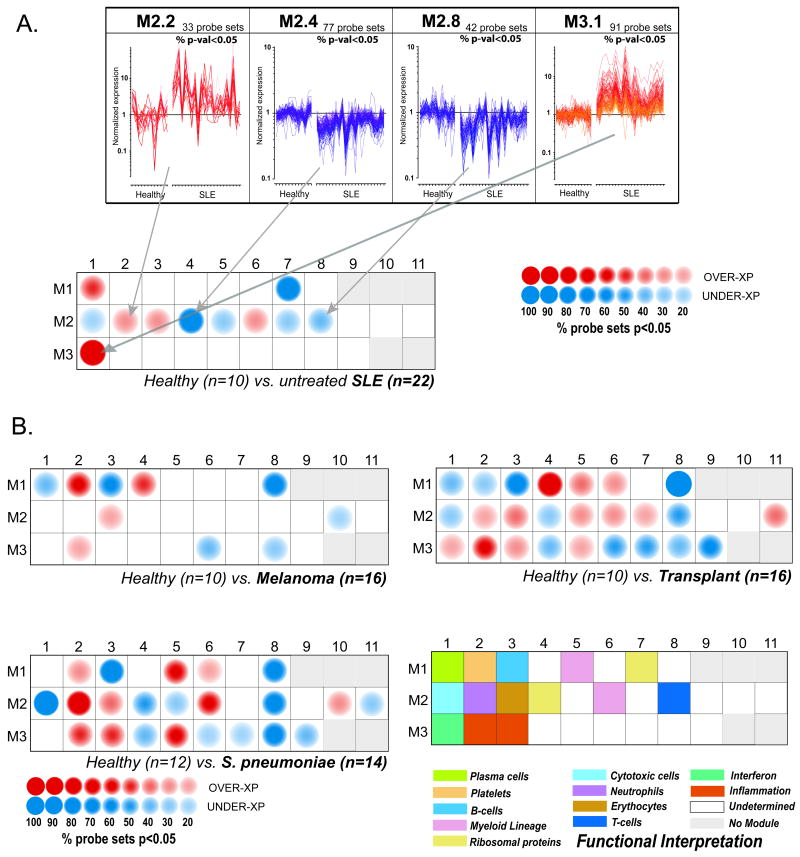
References
-
- Liu K, Mohan C. What do mouse models teach us about human SLE? Clin Immunol. 2006;119:123–30. - PubMed
-
- Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36:1118–25. - PubMed
-
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- ARO54083-01CORT/PHS HHS/United States
- P50 AR055503/AR/NIAMS NIH HHS/United States
- R01-CA078846/CA/NCI NIH HHS/United States
- R01 AR050770/AR/NIAMS NIH HHS/United States
- U01 AI082110/AI/NIAID NIH HHS/United States
- U19-A1057234/PHS HHS/United States
- U19 AI057234/AI/NIAID NIH HHS/United States
- R01 CA078846/CA/NCI NIH HHS/United States
- U19 AI082715/AI/NIAID NIH HHS/United States
- U19-AI082715-01/AI/NIAID NIH HHS/United States
- U01AI082110/AI/NIAID NIH HHS/United States
- P50 AR054083/AR/NIAMS NIH HHS/United States
- ARO55503-01CORT/PHS HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
