

An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p
- PMID: 20193759
- PMCID: PMC2872486
- DOI: 10.1016/j.cellsig.2010.02.008
An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p
Abstract
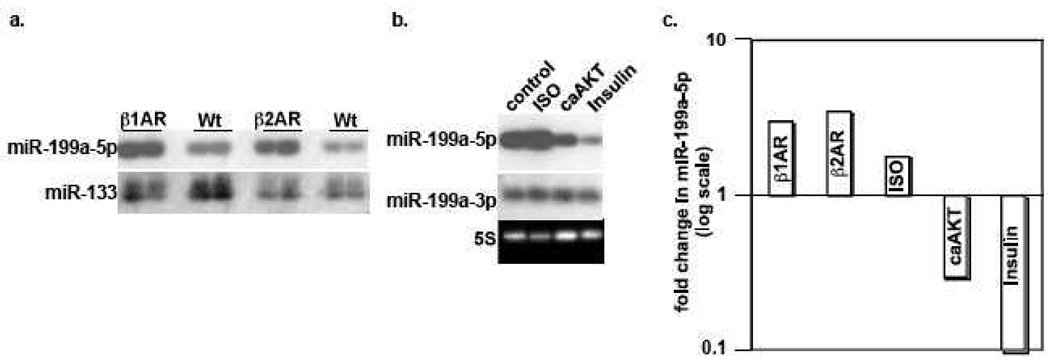
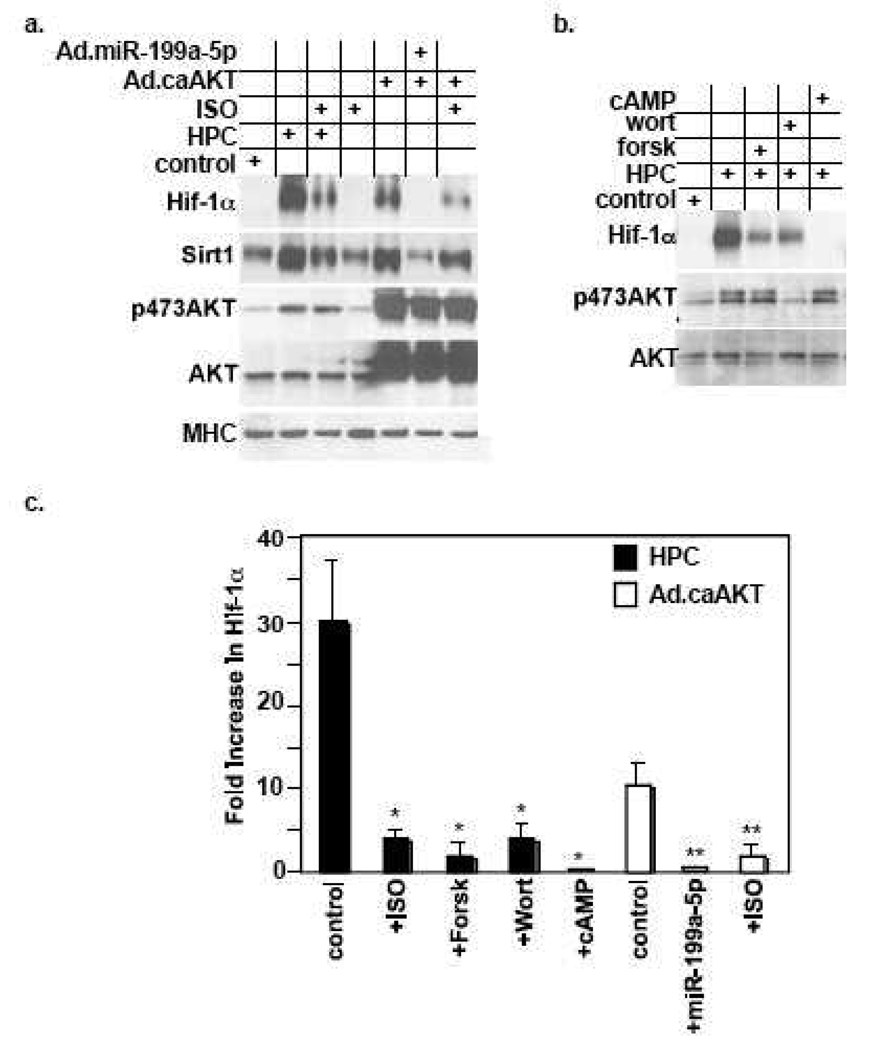
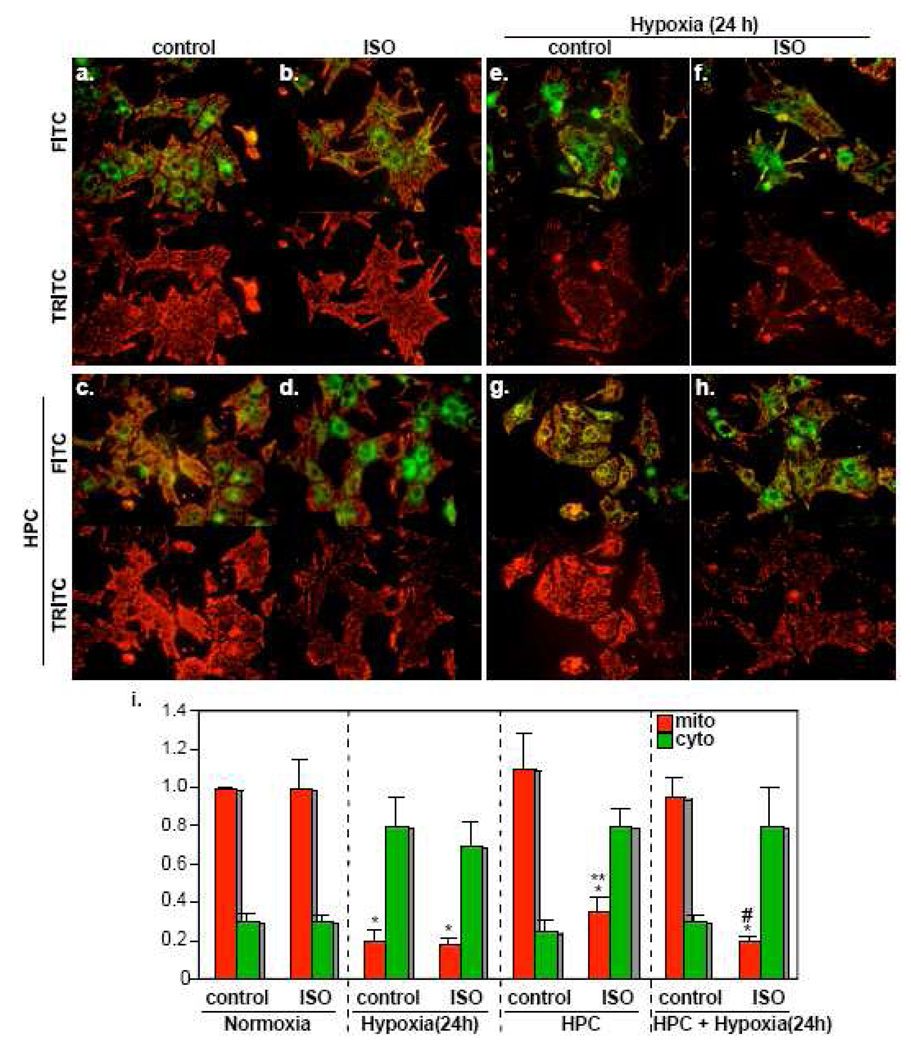
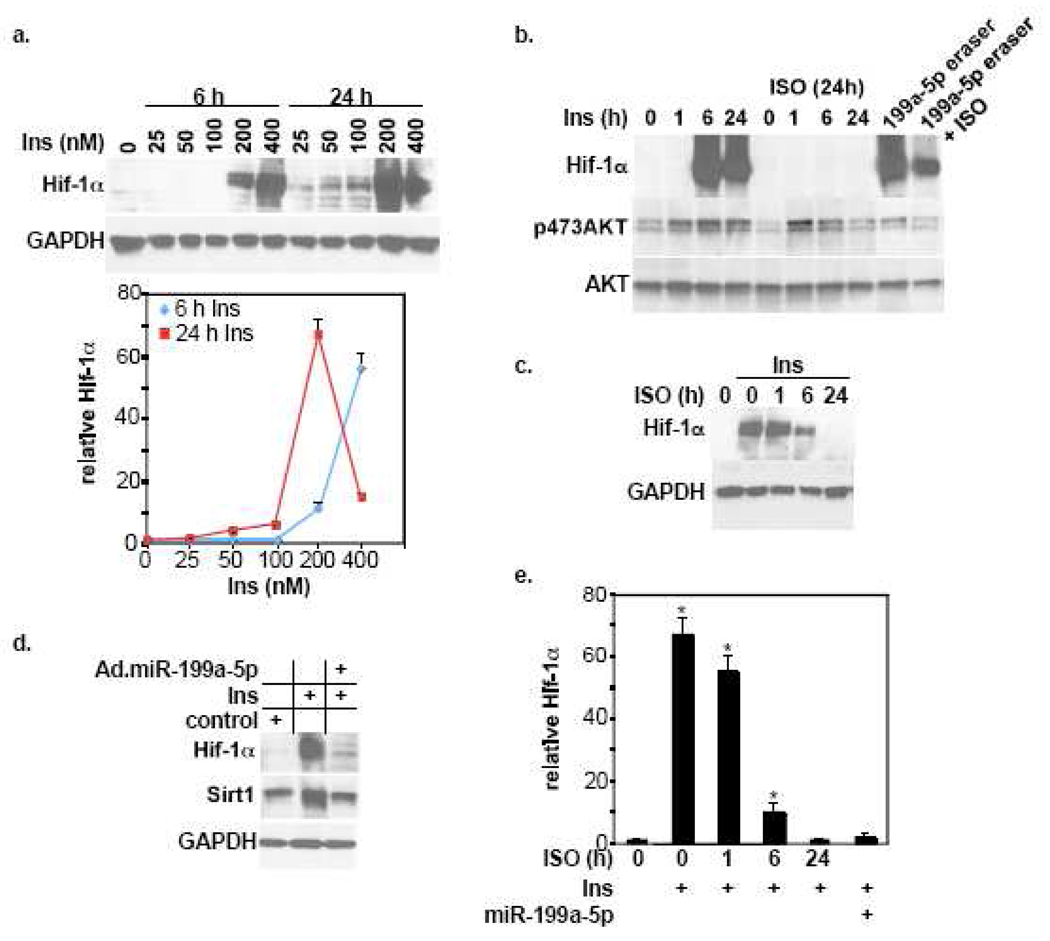
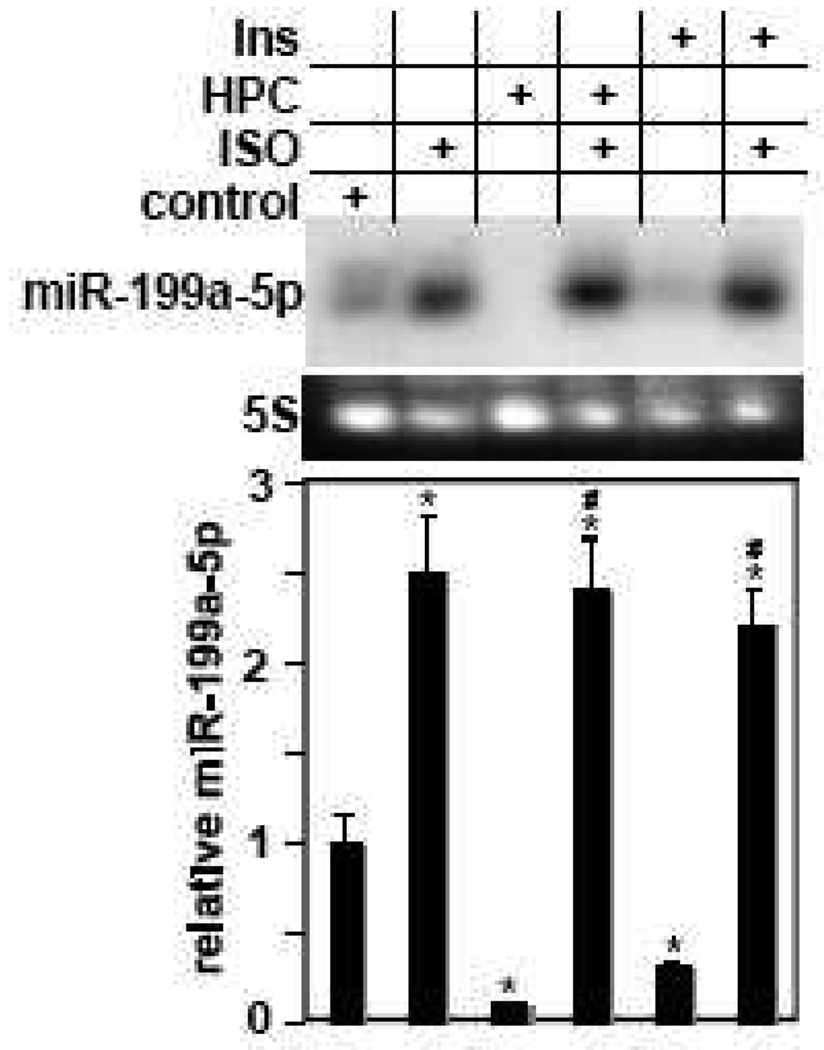
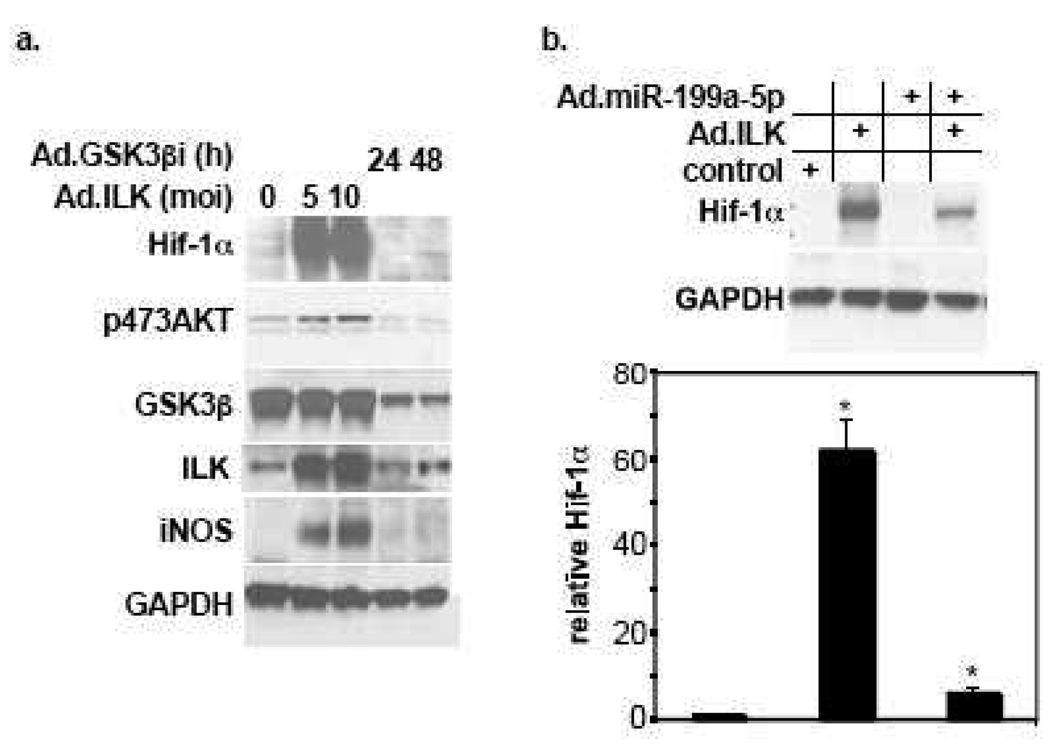
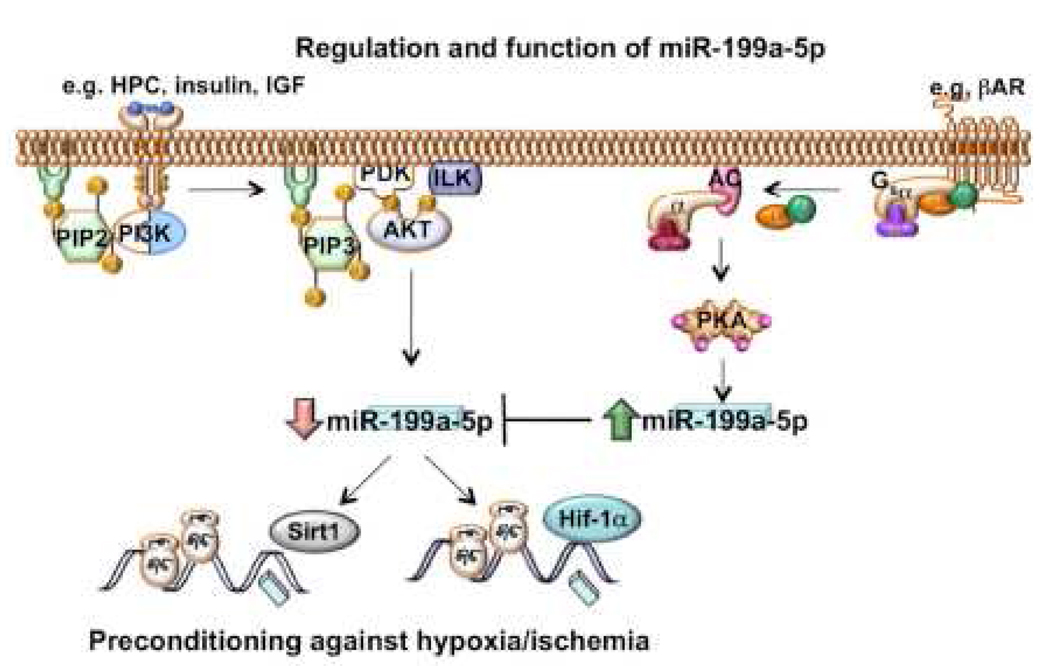
We have recently reported that downregulation of miR-199a-5p is necessary and sufficient for inducing upregulation of its targets, including hypoxia-inducible factor-1 alpha (Hif-1 alpha) and Sirt1, during hypoxia preconditioning (HPC). Conversely, others and we have reported that miR-199a-5p is upregulated during cardiac hypertrophy. Thus, the objective of this study was to delineate the signaling pathways that regulate the expression of miR-199a-5p and its targets, and their role in myocyte survival during hypoxia. Since HPC is mediated through activation of the AKT pathway, we questioned if AKT is sufficient for inducing downregulation of miR-199a-5p. Our present study shows that overexpression of a constitutively active AKT (caAKT) induced 70% reduction in miR-199a-5p and was associated with a robust increase in HiF-1 alpha (10+/-2 fold) and Sirt1 (4+/-0.8 fold) that was reversed by overexpression of miR-199a-5p. Similarly, insulin receptor-stimulated activation of the AKT pathway induced downregulation of miR-199a-5p and upregulation of its targets. In contrast, beta-adrenergic receptor (beta AR) activation in vitro and in vivo, induced 1.8-3.5-fold increase in miR-199a-5p. Accordingly, we predicted that beta AR would antagonize AKT-induced, miR-199a-5p-dependent, upregulation of Hif-1 alpha and Sirt1. Indeed, pre-treating the myocytes with isoproterenol before applying HPC, caAKT, or insulin resulted in 87+/-3%, 75+/-15%, and 100% reductions in Hif-1 alpha expression, respectively, and sensitized the cells to hypoxic injury. Thus, activation of beta-adrenergic signaling counteracts the survival effects of the AKT pathway via upregulating miR-199a-5p.
(c) 2010 Elsevier Inc. All rights reserved.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
