

Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway
- PMID: 20194434
- PMCID: PMC2841333
- DOI: 10.1101/gad.1873910
Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway
Abstract
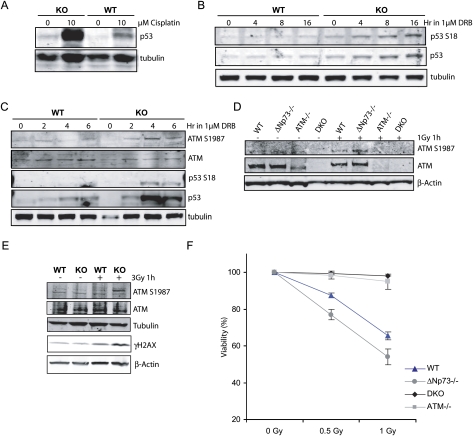
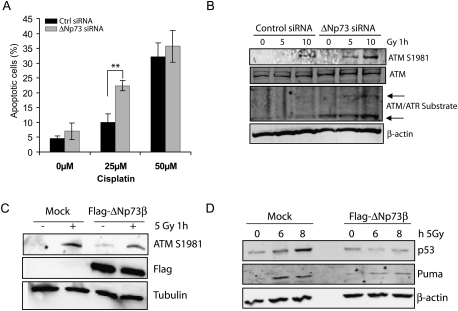
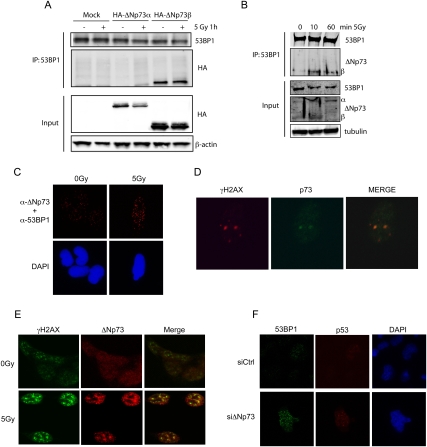
Mice with a complete deficiency of p73 have severe neurological and immunological defects due to the absence of all TAp73 and DeltaNp73 isoforms. As part of our ongoing program to distinguish the biological functions of these isoforms, we generated mice that are selectively deficient for the DeltaNp73 isoform. Mice lacking DeltaNp73 (DeltaNp73(-/-) mice) are viable and fertile but display signs of neurodegeneration. Cells from DeltaNp73(-/-) mice are sensitized to DNA-damaging agents and show an increase in p53-dependent apoptosis. When analyzing the DNA damage response (DDR) in DeltaNp73(-/-) cells, we discovered a completely new role for DeltaNp73 in inhibiting the molecular signal emanating from a DNA break to the DDR pathway. We found that DeltaNp73 localizes directly to the site of DNA damage, can interact with the DNA damage sensor protein 53BP1, and inhibits ATM activation and subsequent p53 phosphorylation. This novel finding may explain why human tumors with high levels of DeltaNp73 expression show enhanced resistance to chemotherapy.
Figures
Comment in
-
{Delta}Np73{beta} puts the brakes on DNA repair.Genes Dev. 2010 Mar 15;24(6):517-20. doi: 10.1101/gad.1914210. Genes Dev. 2010. PMID: 20231313 Free PMC article.
Similar articles
-
Quercetin abrogates chemoresistance in melanoma cells by modulating deltaNp73.BMC Cancer. 2010 Jun 11;10:282. doi: 10.1186/1471-2407-10-282. BMC Cancer. 2010. PMID: 20540768 Free PMC article.
-
STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage.J Cell Sci. 2005 Apr 15;118(Pt 8):1629-39. doi: 10.1242/jcs.01728. Epub 2005 Mar 22. J Cell Sci. 2005. Retraction in: J Cell Sci. 2015 Mar 1;128(5):1064. doi: 10.1242/jcs.169243. PMID: 15784679 Retracted.
-
Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2.Proc Natl Acad Sci U S A. 2010 Jul 20;107(29):12877-82. doi: 10.1073/pnas.0911828107. Epub 2010 Jul 6. Proc Natl Acad Sci U S A. 2010. PMID: 20615966 Free PMC article.
-
Negative autoregulation of p73 and p53 by DeltaNp73 in regulating differentiation and survival of human neuroblastoma cells.Cancer Lett. 2003 Jul 18;197(1-2):105-9. doi: 10.1016/s0304-3835(03)00090-9. Cancer Lett. 2003. PMID: 12880968 Review.
-
p73 induces apoptosis by different mechanisms.Biochem Biophys Res Commun. 2005 Jun 10;331(3):713-7. doi: 10.1016/j.bbrc.2005.03.156. Biochem Biophys Res Commun. 2005. PMID: 15865927 Review.
Cited by
-
p53 and its isoforms in DNA double-stranded break repair.J Zhejiang Univ Sci B. 2019 Jun;20(6):457-466. doi: 10.1631/jzus.B1900167. J Zhejiang Univ Sci B. 2019. PMID: 31090271 Free PMC article. Review.
-
Mechanisms of Drug Resistance and Use of Nanoparticle Delivery to Overcome Resistance in Breast Cancers.Adv Exp Med Biol. 2021;1347:163-181. doi: 10.1007/5584_2021_648. Adv Exp Med Biol. 2021. PMID: 34287795
-
Reviving the guardian of the genome: Small molecule activators of p53.Pharmacol Ther. 2017 Oct;178:92-108. doi: 10.1016/j.pharmthera.2017.03.013. Epub 2017 Mar 27. Pharmacol Ther. 2017. PMID: 28351719 Free PMC article. Review.
-
p73 as a Tissue Architect.Front Cell Dev Biol. 2021 Jul 23;9:716957. doi: 10.3389/fcell.2021.716957. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34368167 Free PMC article. Review.
-
Tissue-specific expression of p73 and p63 isoforms in human tissues.Cell Death Dis. 2021 Jul 27;12(8):745. doi: 10.1038/s41419-021-04017-8. Cell Death Dis. 2021. PMID: 34315849 Free PMC article.
References
-
- Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67:11696–11703. - PubMed
-
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. - PubMed
-
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous