

A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity
- PMID: 20194437
- PMCID: PMC2827839
- DOI: 10.1101/gad.1884910
A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity
Abstract
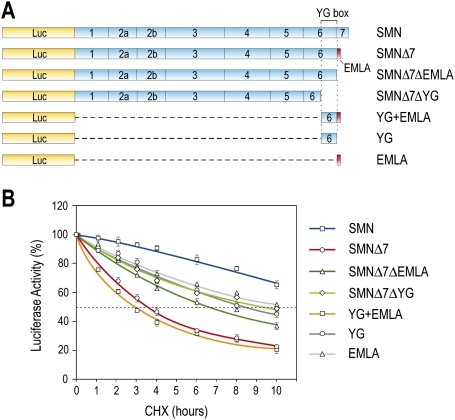
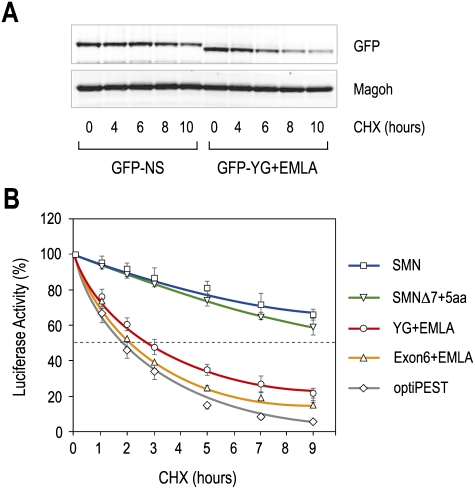
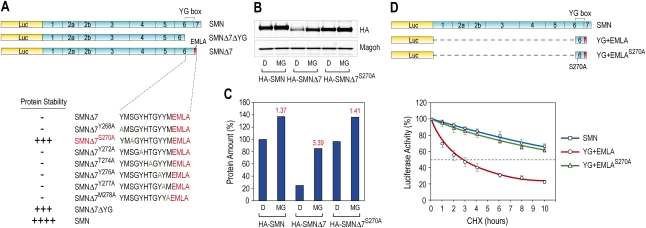
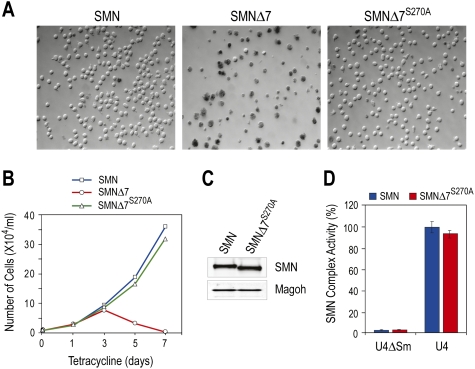
Spinal muscular atrophy (SMA) is caused by homozygous survival of motor neurons 1 (SMN1) gene deletions, leaving a duplicate gene, SMN2, as the sole source of SMN protein. However, most of the mRNA produced from SMN2 pre-mRNA is exon 7-skipped ( approximately 80%), resulting in a highly unstable and almost undetectable protein (SMNDelta7). We show that this splicing defect creates a potent degradation signal (degron; SMNDelta7-DEG) at SMNDelta7's C-terminal 15 amino acids. The S270A mutation inactivates SMNDelta7-DEG, generating a stable SMNDelta7 that rescues viability of SMN-deleted cells. These findings explain a key aspect of the SMA disease mechanism, and suggest new treatment approaches based on interference with SMNDelta7-DEG activity.
Figures
References
-
- Burnett BG, Crawford TO, Sumner CJ. Emerging treatment options for spinal muscular atrophy. Curr Treat Options Neurol. 2009a;11:90–101. - PubMed
-
- Chang HC, Hung WC, Chuang YJ, Jong YJ. Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem Int. 2004;45:1107–1112. - PubMed
-
- Cusco I, Barcelo MJ, Rojas-Garcia R, Illa I, Gamez J, Cervera C, Pou A, Izquierdo G, Baiget M, Tizzano EF. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. 2006;253:21–25. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical