

Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes
- PMID: 20194621
- PMCID: PMC2863588
- DOI: 10.1128/MCB.01614-09
Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes
Erratum in
-
Correction for Li and Guan, "Human Mitochondrial Leucyl-tRNA Synthetase Corrects Mitochondrial Dysfunctions Due to the tRNALeu(UUR) A3243G Mutation, Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Symptoms and Diabetes".Mol Cell Biol. 2017 Aug 28;37(18):e00335-17. doi: 10.1128/MCB.00335-17. Print 2017 Sep 15. Mol Cell Biol. 2017. PMID: 28847973 Free PMC article. No abstract available.
Abstract
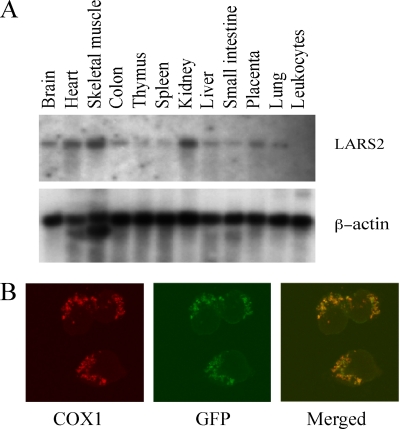
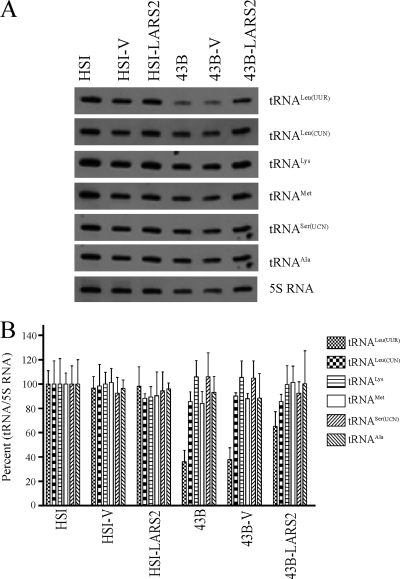
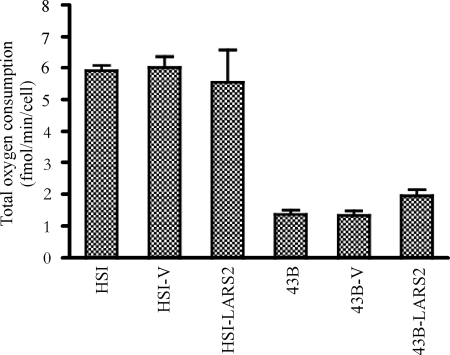
Mutations in mitochondrial tRNA genes are associated with a wide spectrum of human diseases. In particular, the tRNA(Leu(UUR)) A3243G mutation causes mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms (MELAS) and 2% of cases of type 2 diabetes. The primary defect in this mutation was an inefficient aminoacylation of the tRNA(Leu(UUR)). In the present study, we have investigated the molecular mechanism of the A3243G mutation and whether the overexpression of human mitochondrial leucyl-tRNA synthetase (LARS2) in the cytoplasmic hybrid (cybrid) cells carrying the A3243G mutation corrects the mitochondrial dysfunctions. Human LARS2 localizes exclusively to mitochondria, and LARS2 is expressed ubiquitously but most abundantly in tissues with high metabolic rates. We showed that the alteration of aminoacylation tRNA(Leu(UUR)) caused by the A3243G mutation led to mitochondrial translational defects and thereby reduced the aminoacylated efficiencies of tRNA(Leu(UUR)) as well as tRNA(Ala) and tRNA(Met). We demonstrated that the transfer of human mitochondrial leucyl-tRNA synthetase into the cybrid cells carrying the A3243G mutation improved the efficiency of aminoacylation and stability of mitochondrial tRNAs and then increased the rates of mitochondrial translation and respiration, consequently correcting the mitochondrial dysfunction. These findings provide new insights into the molecular mechanism of maternally inherited diseases and a step toward therapeutic interventions for these disorders.
Figures




Similar articles
-
The pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR) decreases the efficiency of aminoacylation.Biochemistry. 2003 Feb 4;42(4):958-64. doi: 10.1021/bi026882r. Biochemistry. 2003. PMID: 12549915
-
Recognition of human mitochondrial tRNALeu(UUR) by its cognate leucyl-tRNA synthetase.J Mol Biol. 2004 May 21;339(1):17-29. doi: 10.1016/j.jmb.2004.03.066. J Mol Biol. 2004. PMID: 15123417
-
The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes.J Biol Chem. 2000 Jun 23;275(25):19198-209. doi: 10.1074/jbc.M908734199. J Biol Chem. 2000. PMID: 10858457
-
Molecular pathology of MELAS and L-arginine effects.Biochim Biophys Acta. 2012 May;1820(5):608-14. doi: 10.1016/j.bbagen.2011.09.005. Epub 2011 Sep 14. Biochim Biophys Acta. 2012. PMID: 21944974 Review.
-
Early onset cardiomyopathy associated with the mitochondrial tRNALeu((UUR)) 3271T>C MELAS mutation.Biochem Biophys Res Commun. 2015 Mar 13;458(3):601-604. doi: 10.1016/j.bbrc.2015.01.157. Epub 2015 Feb 11. Biochem Biophys Res Commun. 2015. PMID: 25680467 Review.
Cited by
-
Screening of mitochondrial mutations and insertion-deletion polymorphism in gestational diabetes mellitus in the Asian Indian population.Saudi J Biol Sci. 2015 May;22(3):243-8. doi: 10.1016/j.sjbs.2014.11.001. Epub 2014 Nov 12. Saudi J Biol Sci. 2015. PMID: 25972744 Free PMC article.
-
The Role and Research Progress of Mitochondria in Sensorineural Hearing Loss.Mol Neurobiol. 2025 Jun;62(6):6913-6921. doi: 10.1007/s12035-024-04470-4. Epub 2024 Sep 18. Mol Neurobiol. 2025. PMID: 39292339 Free PMC article. Review.
-
Loss of the mitochondrial protein-only ribonuclease P complex causes aberrant tRNA processing and lethality in Drosophila.Nucleic Acids Res. 2016 Jul 27;44(13):6409-22. doi: 10.1093/nar/gkw338. Epub 2016 Apr 30. Nucleic Acids Res. 2016. PMID: 27131785 Free PMC article.
-
tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors.Int J Mol Sci. 2021 Jan 6;22(2):496. doi: 10.3390/ijms22020496. Int J Mol Sci. 2021. PMID: 33419045 Free PMC article. Review.
-
The protein-only RNase Ps, endonucleases that cleave pre-tRNA: Biological relevance, molecular architectures, substrate recognition and specificity, and protein interactomes.Wiley Interdiscip Rev RNA. 2024 Mar-Apr;15(2):e1836. doi: 10.1002/wrna.1836. Wiley Interdiscip Rev RNA. 2024. PMID: 38453211 Free PMC article.
References
-
- Börner, G. V., M. Zeviani, V. Tiranti, F. Carrara, S. Hoffmann, K. D. Gerbitz, H. Lochmüller, D. Pongratz, T. Klopstock, A. Melberg, E. Holme, and S. Pääbo. 2000. Decreased aminoacylation of mutant tRNAs in MELAS but not in MERRF patients. Hum. Mol. Genet. 9:467-475. - PubMed
-
- Bullard, J. M., Y. C. Cai, and L. L. Spremulli. 2000. Expression and characterization of the human mitochondrial leucyl-tRNA synthetase. Biochim. Biophys. Acta 1490:245-258. - PubMed
-
- Chomyn, A. 1996. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 264:197-211. - PubMed
-
- Chomyn, A., J. A. Enriquez, V. Micol, P. Fernandez-Silva, and G. Attardi. 2000. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 275:19198-19209. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous