

Allele-specific tumor spectrum in pten knockin mice
- PMID: 20194734
- PMCID: PMC2841921
- DOI: 10.1073/pnas.0912524107
Allele-specific tumor spectrum in pten knockin mice
Abstract
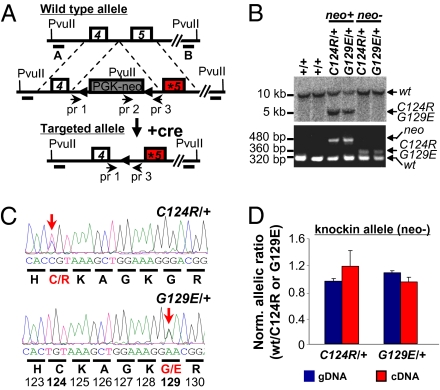
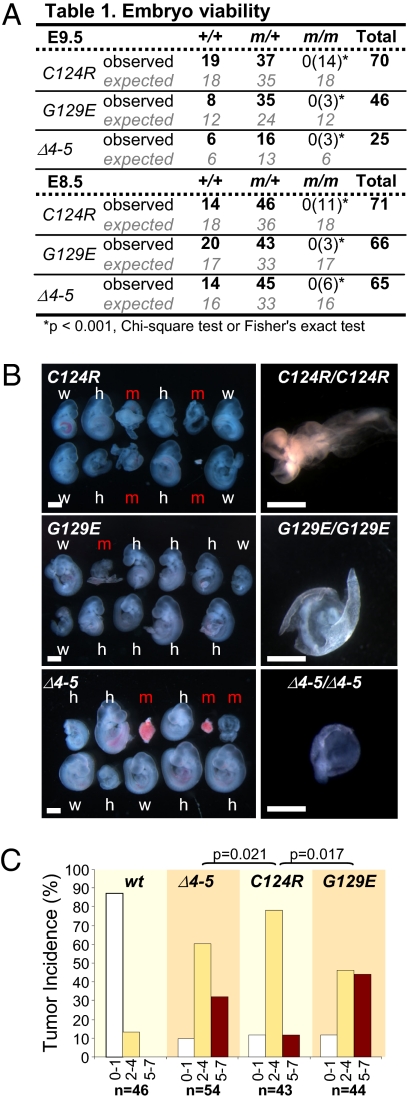
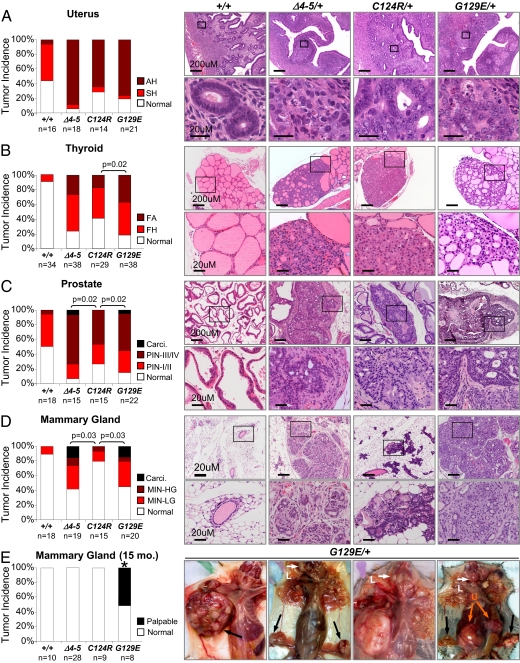
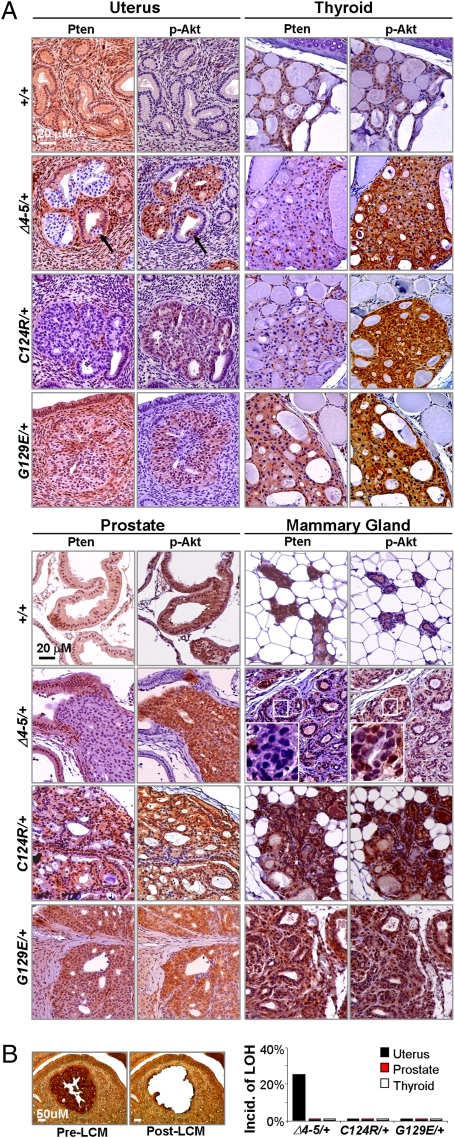
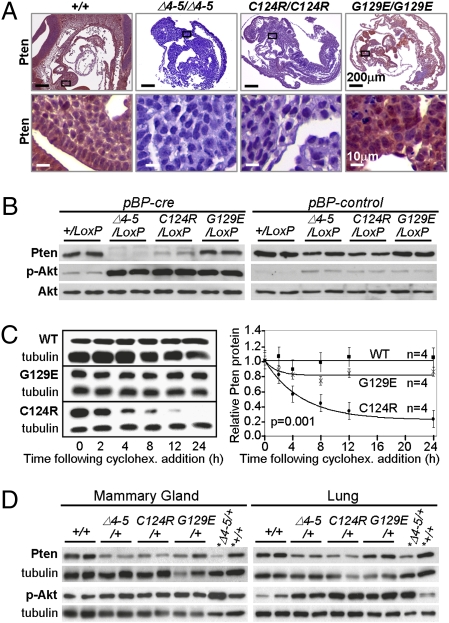
Germline mutations in the tumor suppressor gene PTEN (phosphatase and tensin homology deleted on chromosome 10) cause Cowden and Bannayan-Riley-Ruvalcaba (BRR) syndromes, two dominantly inherited disorders characterized by mental retardation, multiple hamartomas, and variable cancer risk. Here, we modeled three sentinel mutant alleles of PTEN identified in patients with Cowden syndrome and show that the nonsense Pten(4-5) and missense Pten(C124R) and Pten(G129E) alleles lacking lipid phosphatase activity cause similar developmental abnormalities but distinct tumor spectra with varying severity and age of onset. Allele-specific differences may be accounted for by loss of function for Pten(4-5), hypomorphic function for Pten(C124R), and gain of function for Pten(G129E). These data demonstrate that the variable tumor phenotypes observed in patients with Cowden and BRR syndromes can be attributed to specific mutations in PTEN that alter protein function through distinct mechanisms.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. - PubMed
-
- Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. - PubMed
-
- Liaw D, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
