

Large-scale conformational sampling of proteins using temperature-accelerated molecular dynamics
- PMID: 20194785
- PMCID: PMC2841907
- DOI: 10.1073/pnas.0914540107
Large-scale conformational sampling of proteins using temperature-accelerated molecular dynamics
Abstract
We show how to apply the method of temperature-accelerated molecular dynamics (TAMD) in collective variables [Maragliano L, Vanden-Eijnden E (2006) Chem Phys Lett 426:168-175] to sample the conformational space of multidomain proteins in all-atom, explicitly solvated molecular dynamics simulations. The method allows the system to hyperthermally explore the free-energy surface in a set of collective variables computed at the physical temperature. As collective variables, we pick Cartesian coordinates of centers of contiguous subdomains. The method is applied to the GroEL subunit, a 55-kDa, three-domain protein, and HIV-1 gp120. For GroEL, the method induces in about 40 ns conformational changes that recapitulate the t --> r('') transition and are not observed in unaccelerated molecular dynamics: The apical domain is displaced by 30 A, with a twist of 90 degrees relative to the equatorial domain, and the root-mean-squared deviation relative to the r('') conformer is reduced from 13 to 5 A, representing fairly high predictive capability. For gp120, the method predicts both counterrotation of inner and outer domains and disruption of the so-called bridging sheet. In particular, TAMD on gp120 initially in the CD4-bound conformation visits conformations that deviate by 3.6 A from the gp120 conformer in complex with antibody F105, again reflecting good predictive capability. TAMD generates plausible all-atom models of the so-far structurally uncharacterized unliganded conformation of HIV-1 gp120, which may prove useful in the development of inhibitors and immunogens. The fictitious temperature employed also gives a rough estimate of 10 kcal/mol for the free-energy barrier between conformers in both cases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
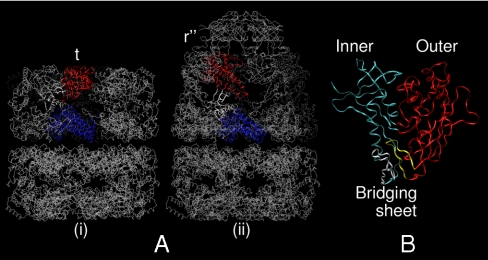
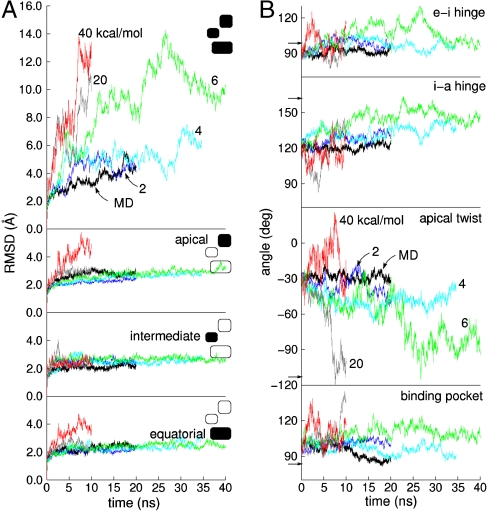
 as labeled in the Uppermost panel. (B) ICs used to measure conformation derived from the CVs vs. simulation time. From Top to Bottom are shown the hinge angle between the equatorial and intermediate domains, the hinge angle between intermediate and apical domains, the dihedral describing the twist of the apical domain, and the angle defining the disposition of the nucleotide binding pocket. Color scheme is same as in A. Arrows along the y axis denote IC values in the crystallographic r′′ state (42).
as labeled in the Uppermost panel. (B) ICs used to measure conformation derived from the CVs vs. simulation time. From Top to Bottom are shown the hinge angle between the equatorial and intermediate domains, the hinge angle between intermediate and apical domains, the dihedral describing the twist of the apical domain, and the angle defining the disposition of the nucleotide binding pocket. Color scheme is same as in A. Arrows along the y axis denote IC values in the crystallographic r′′ state (42).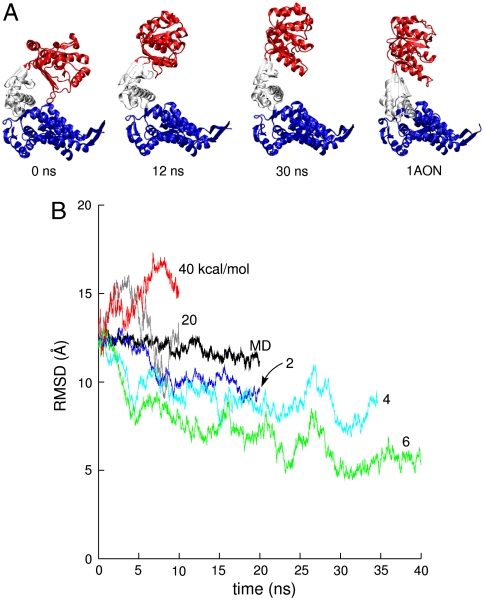
 . Rightmost panel shows the crystallographic r′′ state from the same viewpoint. (B) Whole-subunit rmsd relative to the crystallographic r′′ state (42) for traditional MD simulation and various TAMD simulations. Color scheme follows that of Fig. 2.
. Rightmost panel shows the crystallographic r′′ state from the same viewpoint. (B) Whole-subunit rmsd relative to the crystallographic r′′ state (42) for traditional MD simulation and various TAMD simulations. Color scheme follows that of Fig. 2.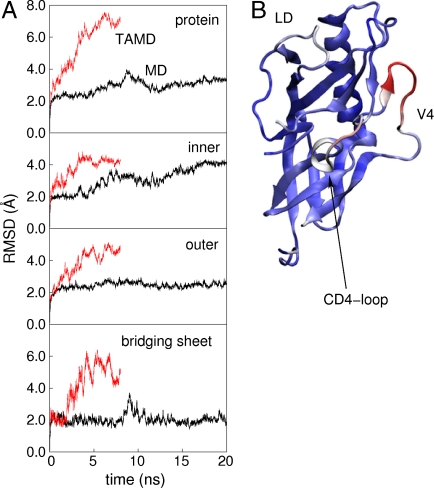
 , followed by 4 ns of unaccelerated MD. (B) Cartoon rendering of the HIV-1 gp120 outer domain colorized according to the difference in per-residue rms fluctuation in TAMD vs MD, with warmer colors signifying larger differences. The D-loop (“LD”), V4 loop, and CD4-binding loop are indicated.
, followed by 4 ns of unaccelerated MD. (B) Cartoon rendering of the HIV-1 gp120 outer domain colorized according to the difference in per-residue rms fluctuation in TAMD vs MD, with warmer colors signifying larger differences. The D-loop (“LD”), V4 loop, and CD4-binding loop are indicated.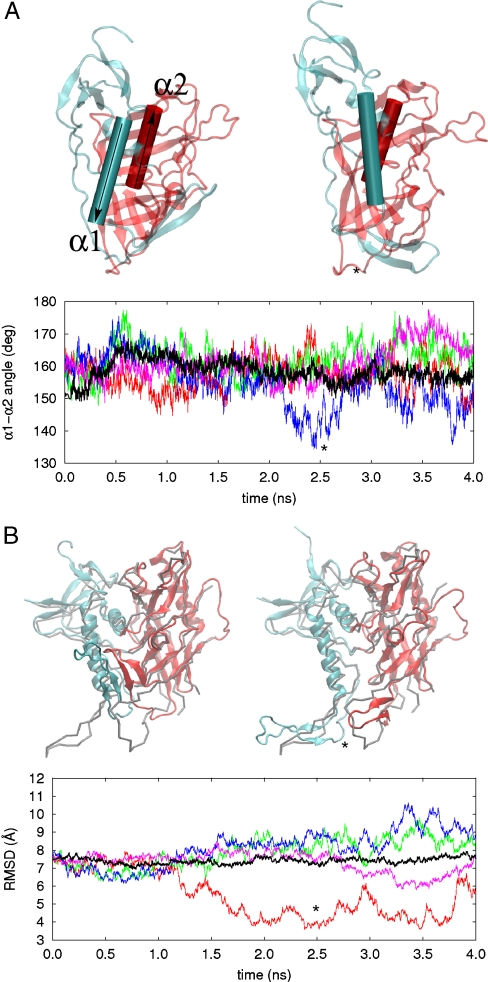
References
-
- Bennett WS, Huber R, Engel J. Structural and functional aspects of domain motion in proteins. Crit Rev Biochem. 1984;15:291–384. - PubMed
-
- Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6748. - PubMed
-
- Schulz GE. Domain motion in proteins. Curr Opin Struct Biol. 1991;1:883–888.
-
- Hayward S, Go N. Collective variable description of native protein dynamics. Annu Rev Phys Chem. 1995;46:223–50. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
