

Improved survival of children and adolescents with sickle cell disease
- PMID: 20194891
- PMCID: PMC2867259
- DOI: 10.1182/blood-2009-07-233700
Improved survival of children and adolescents with sickle cell disease
Abstract
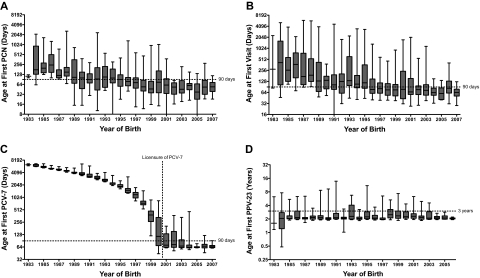
The survival of young children with sickle cell disease (SCD) has improved, but less is known about older children and adolescents. We studied the Dallas Newborn Cohort (DNC) to estimate contemporary 18-year survival for newborns with SCD and document changes in the causes and ages of death over time. We also explored whether improvements in the quality of medical care were temporally associated with survival. The DNC now includes 940 subjects with 8857 patient-years of follow-up. Most children with sickle cell anemia (93.9%) and nearly all children with milder forms of SCD (98.4%) now live to become adults. The incidence of death and the pattern of mortality changed over the duration of the cohort. Sepsis is no longer the leading cause of death. All the recent deaths in the cohort occurred in patients 18 years or older, most shortly after the transition to adult care. Quality of care in the DNC has improved over time, with significantly more timely initial visits and preventive interventions for young children. In summary, most children with SCD now survive the childhood years, but young adults who transition to adult medical care are at high risk for early death.
Figures
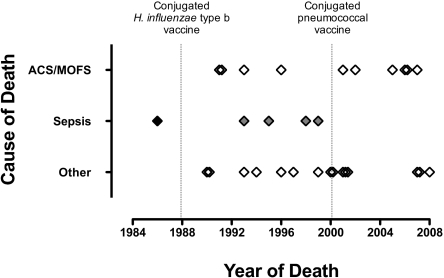
 ) or Streptococcus pneumoniae sepsis (
) or Streptococcus pneumoniae sepsis ( ) since the availability of the protein-conjugate vaccine against either bacterium (dotted lines).
) since the availability of the protein-conjugate vaccine against either bacterium (dotted lines).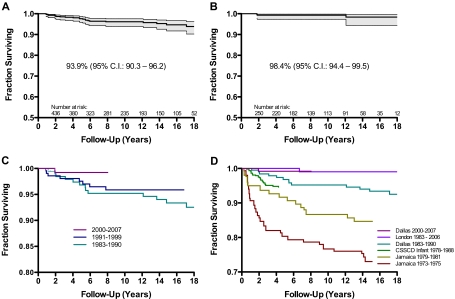
References
-
- Powars DR, Hiti A, Ramicone E, Johnson C, Chan L. Outcome in hemoglobin SC disease: a four-decade observational study of clinical, hematologic, and genetic factors. Am J Hematol. 2002;70(3):206–215. - PubMed
-
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia: a randomized trial. N Engl J Med. 1986;314(25):1593–1599. - PubMed
-
- Vichinsky E, Hurst D, Earles A, Kleman K, Lubin B. Newborn screening for sickle cell disease: effect on mortality. Pediatrics. 1988;81(6):749–755. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
