

Unfolding simulations reveal the mechanism of extreme unfolding cooperativity in the kinetically stable alpha-lytic protease
- PMID: 20195497
- PMCID: PMC2829044
- DOI: 10.1371/journal.pcbi.1000689
Unfolding simulations reveal the mechanism of extreme unfolding cooperativity in the kinetically stable alpha-lytic protease
Abstract
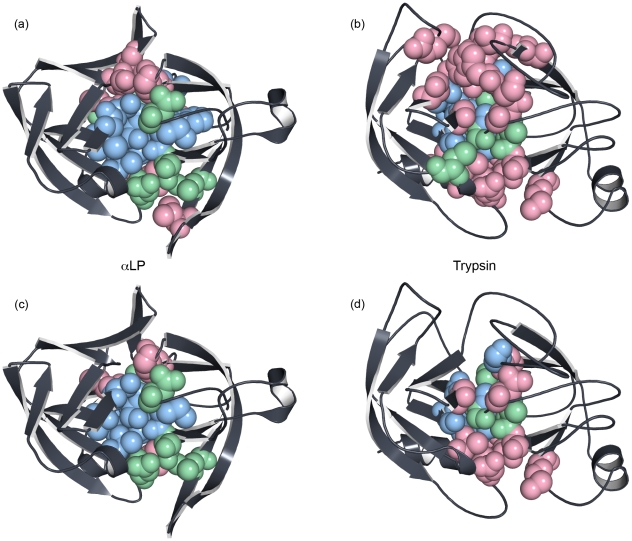
Kinetically stable proteins, those whose stability is derived from their slow unfolding kinetics and not thermodynamics, are examples of evolution's best attempts at suppressing unfolding. Especially in highly proteolytic environments, both partially and fully unfolded proteins face potential inactivation through degradation and/or aggregation, hence, slowing unfolding can greatly extend a protein's functional lifetime. The prokaryotic serine protease alpha-lytic protease (alphaLP) has done just that, as its unfolding is both very slow (t(1/2) approximately 1 year) and so cooperative that partial unfolding is negligible, providing a functional advantage over its thermodynamically stable homologs, such as trypsin. Previous studies have identified regions of the domain interface as critical to alphaLP unfolding, though a complete description of the unfolding pathway is missing. In order to identify the alphaLP unfolding pathway and the mechanism for its extreme cooperativity, we performed high temperature molecular dynamics unfolding simulations of both alphaLP and trypsin. The simulated alphaLP unfolding pathway produces a robust transition state ensemble consistent with prior biochemical experiments and clearly shows that unfolding proceeds through a preferential disruption of the domain interface. Through a novel method of calculating unfolding cooperativity, we show that alphaLP unfolds extremely cooperatively while trypsin unfolds gradually. Finally, by examining the behavior of both domain interfaces, we propose a model for the differential unfolding cooperativity of alphaLP and trypsin involving three key regions that differ between the kinetically stable and thermodynamically stable classes of serine proteases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
The folding landscape of an alpha-lytic protease variant reveals the role of a conserved beta-hairpin in the development of kinetic stability.Proteins. 2005 Oct 1;61(1):105-14. doi: 10.1002/prot.20525. Proteins. 2005. PMID: 16044461
-
The folding landscape of Streptomyces griseus protease B reveals the energetic costs and benefits associated with evolving kinetic stability.Protein Sci. 2004 Feb;13(2):381-90. doi: 10.1110/ps.03336804. Epub 2004 Jan 10. Protein Sci. 2004. PMID: 14718653 Free PMC article.
-
Comprehensive analysis of protein folding activation thermodynamics reveals a universal behavior violated by kinetically stable proteases.J Mol Biol. 2005 Mar 25;347(2):355-66. doi: 10.1016/j.jmb.2005.01.032. Epub 2005 Jan 28. J Mol Biol. 2005. PMID: 15740746
-
Limited cooperativity in protein folding.Curr Opin Struct Biol. 2016 Feb;36:58-66. doi: 10.1016/j.sbi.2015.12.001. Epub 2016 Feb 2. Curr Opin Struct Biol. 2016. PMID: 26845039 Review.
-
How cooperative are protein folding and unfolding transitions?Protein Sci. 2016 Nov;25(11):1924-1941. doi: 10.1002/pro.3015. Epub 2016 Sep 13. Protein Sci. 2016. PMID: 27522064 Free PMC article. Review.
Cited by
-
Propeptides are sufficient to regulate organelle-specific pH-dependent activation of furin and proprotein convertase 1/3.J Mol Biol. 2012 Oct 12;423(1):47-62. doi: 10.1016/j.jmb.2012.06.023. Epub 2012 Jun 25. J Mol Biol. 2012. PMID: 22743102 Free PMC article.
-
Insights into the Importance of DFD-Motif and Insertion I1 in Stabilizing the DFD-Out Conformation of Mnk2 Kinase.ACS Med Chem Lett. 2013 Jun 24;4(8):736-41. doi: 10.1021/ml400145x. eCollection 2013 Aug 8. ACS Med Chem Lett. 2013. PMID: 24900740 Free PMC article.
-
Computational design of a cutinase for plastic biodegradation by mining molecular dynamics simulations trajectories.Comput Struct Biotechnol J. 2022 Jan 5;20:459-470. doi: 10.1016/j.csbj.2021.12.042. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35070168 Free PMC article.
-
The mechanism by which a propeptide-encoded pH sensor regulates spatiotemporal activation of furin.J Biol Chem. 2013 Jun 28;288(26):19154-65. doi: 10.1074/jbc.M112.442681. Epub 2013 May 7. J Biol Chem. 2013. PMID: 23653353 Free PMC article.
-
Interactions with the bifunctional interface of the transcriptional coactivator DCoH1 are kinetically regulated.J Biol Chem. 2015 Feb 13;290(7):4319-29. doi: 10.1074/jbc.M114.616870. Epub 2014 Dec 23. J Biol Chem. 2015. PMID: 25538247 Free PMC article.
References
-
- Sohl JL, Jaswal SS, Agard DA. Unfolded conformations of alpha-lytic protease are more stable than its native state. Nature. 1998;395:817–819. - PubMed
-
- Jaswal SS, Sohl JL, Davis JH, Agard DA. Energetic landscape of alpha-lytic protease optimizes longevity through kinetic stability. Nature. 2002;415:343–346. - PubMed
-
- Jaswal SS, Truhlar SM, Dill KA, Agard DA. Comprehensive analysis of protein folding activation thermodynamics reveals a universal behavior violated by kinetically stable proteases. J Mol Biol. 2005;347:355–366. - PubMed
