

Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags
- PMID: 20195501
- PMCID: PMC2829049
- DOI: 10.1371/journal.pgen.1000862
Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags
Abstract
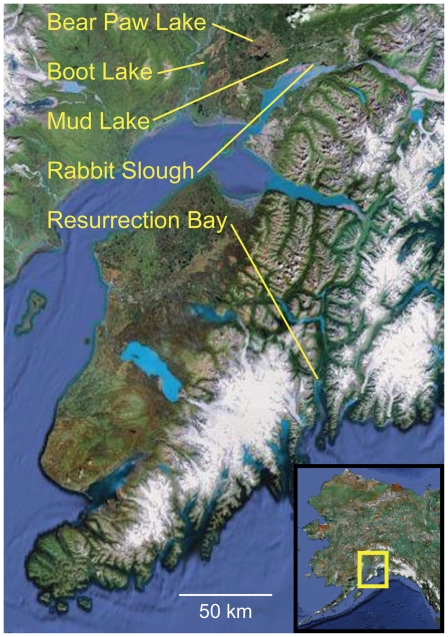
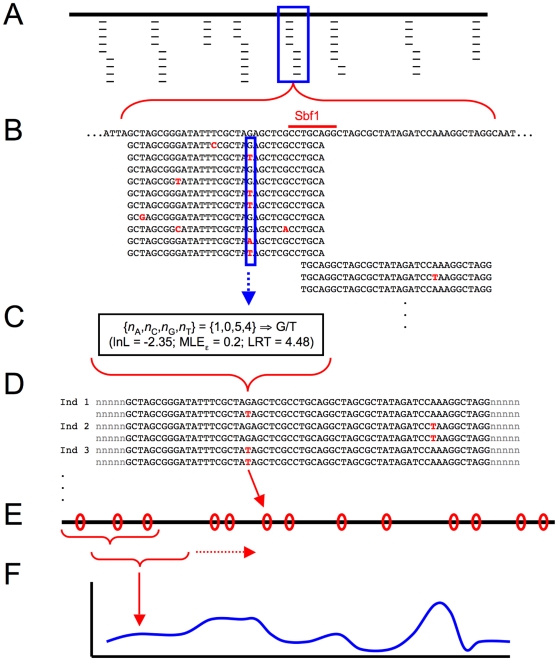
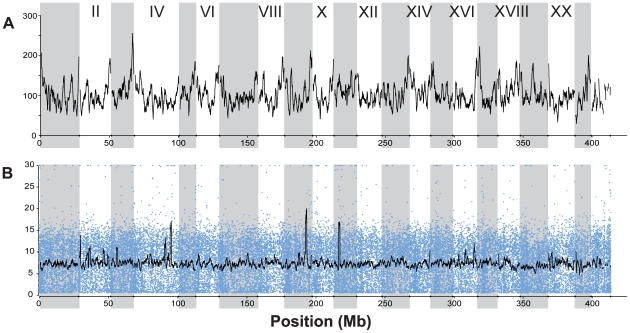
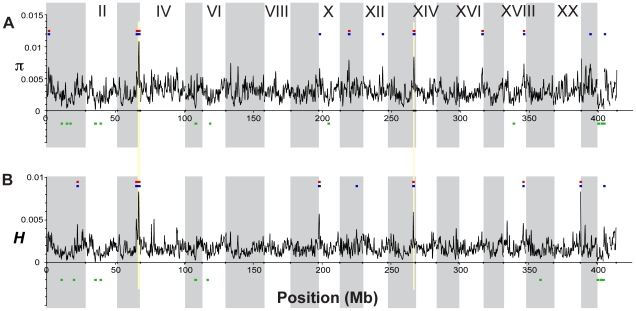
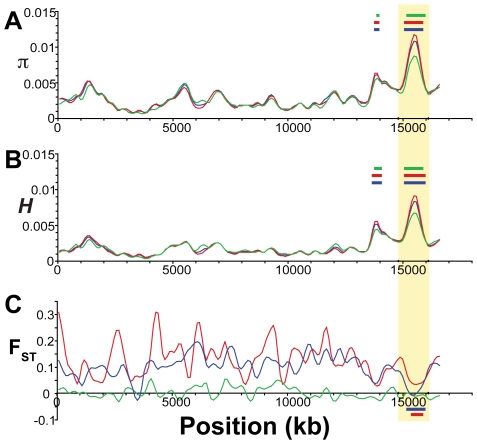
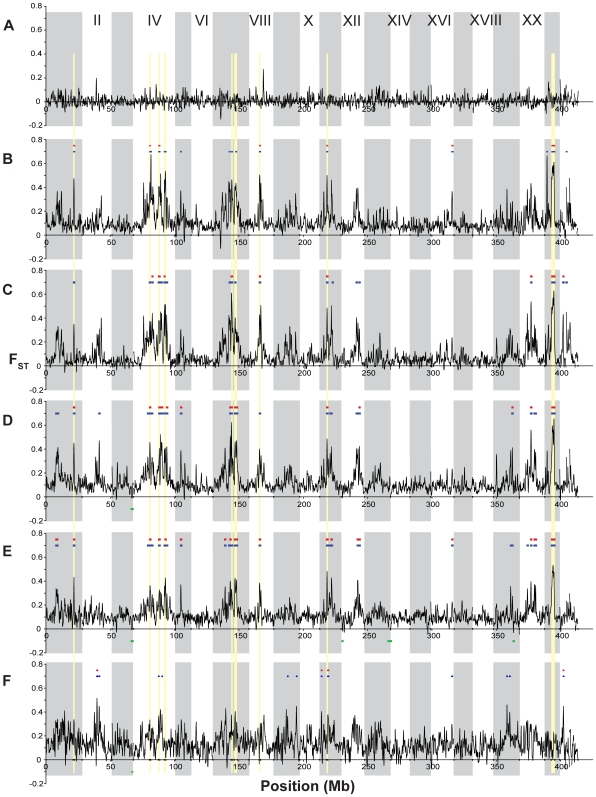
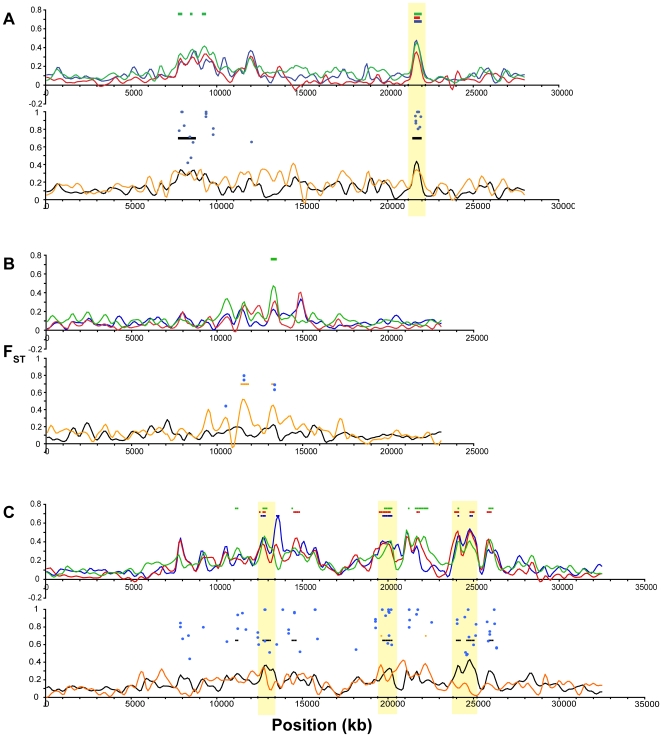
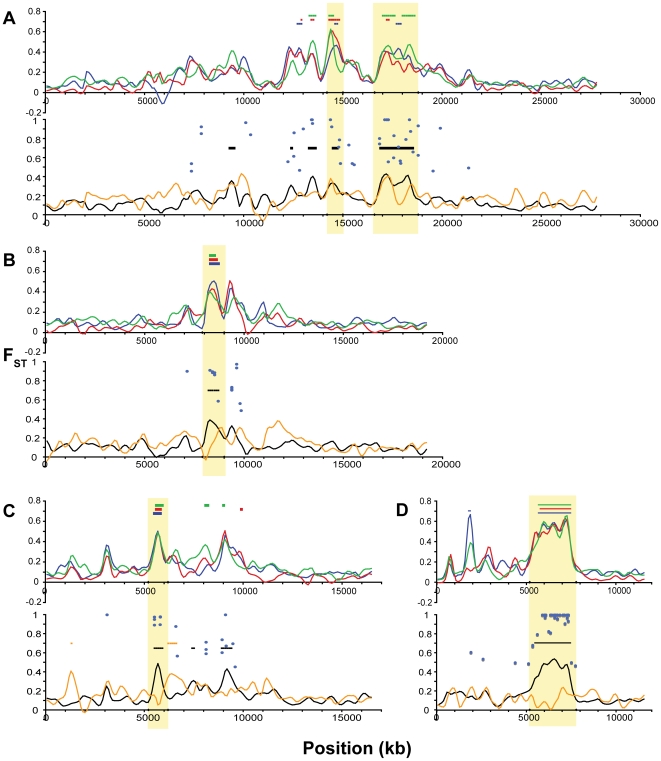
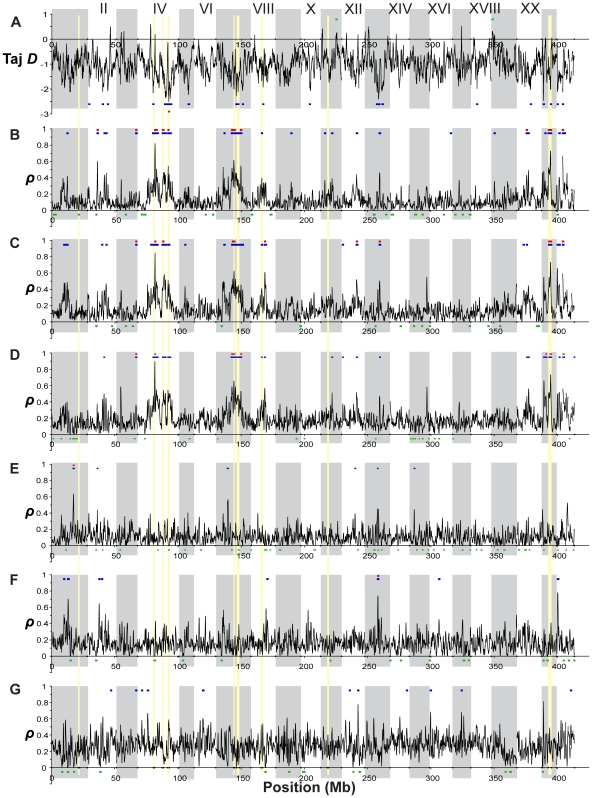
Next-generation sequencing technology provides novel opportunities for gathering genome-scale sequence data in natural populations, laying the empirical foundation for the evolving field of population genomics. Here we conducted a genome scan of nucleotide diversity and differentiation in natural populations of threespine stickleback (Gasterosteus aculeatus). We used Illumina-sequenced RAD tags to identify and type over 45,000 single nucleotide polymorphisms (SNPs) in each of 100 individuals from two oceanic and three freshwater populations. Overall estimates of genetic diversity and differentiation among populations confirm the biogeographic hypothesis that large panmictic oceanic populations have repeatedly given rise to phenotypically divergent freshwater populations. Genomic regions exhibiting signatures of both balancing and divergent selection were remarkably consistent across multiple, independently derived populations, indicating that replicate parallel phenotypic evolution in stickleback may be occurring through extensive, parallel genetic evolution at a genome-wide scale. Some of these genomic regions co-localize with previously identified QTL for stickleback phenotypic variation identified using laboratory mapping crosses. In addition, we have identified several novel regions showing parallel differentiation across independent populations. Annotation of these regions revealed numerous genes that are candidates for stickleback phenotypic evolution and will form the basis of future genetic analyses in this and other organisms. This study represents the first high-density SNP-based genome scan of genetic diversity and differentiation for populations of threespine stickleback in the wild. These data illustrate the complementary nature of laboratory crosses and population genomic scans by confirming the adaptive significance of previously identified genomic regions, elucidating the particular evolutionary and demographic history of such regions in natural populations, and identifying new genomic regions and candidate genes of evolutionary significance.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Fisher RA. New York: Dover; 1958. The Genetical Theory of Natural Selection.
-
- Wright S. Chicago: University of Chicago Press; 1978. Evolution and the genetics of populations.
-
- Kimura M, Ota T. Theoretical aspects of population genetics. Monogr Popul Biol. 1971;4:1–219. - PubMed
-
- Gillespie JH. The status of the Neutral Theory: The Neutral Theory of Molecular Evolution. Science. 1984;224:732–733. - PubMed
-
- Charlesworth B. Effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 2009;10:195–205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
