

Cdk2 is required for p53-independent G2/M checkpoint control
- PMID: 20195506
- PMCID: PMC2829054
- DOI: 10.1371/journal.pgen.1000863
Cdk2 is required for p53-independent G2/M checkpoint control
Abstract
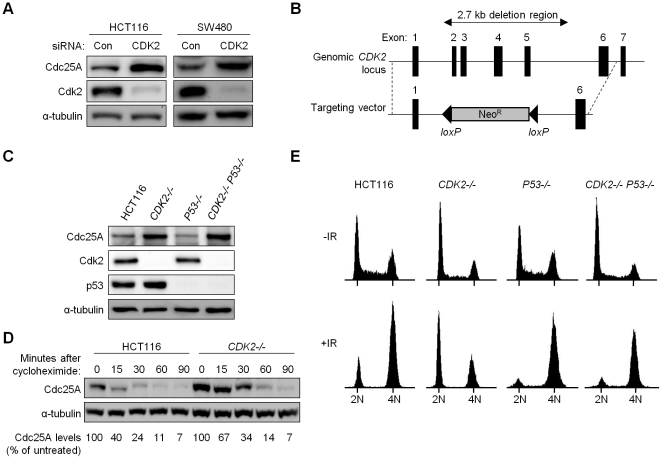
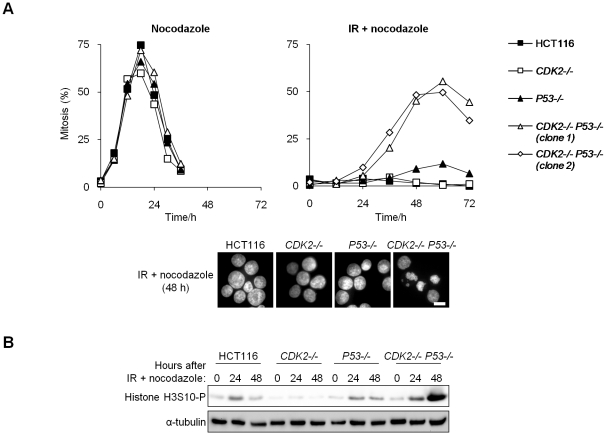
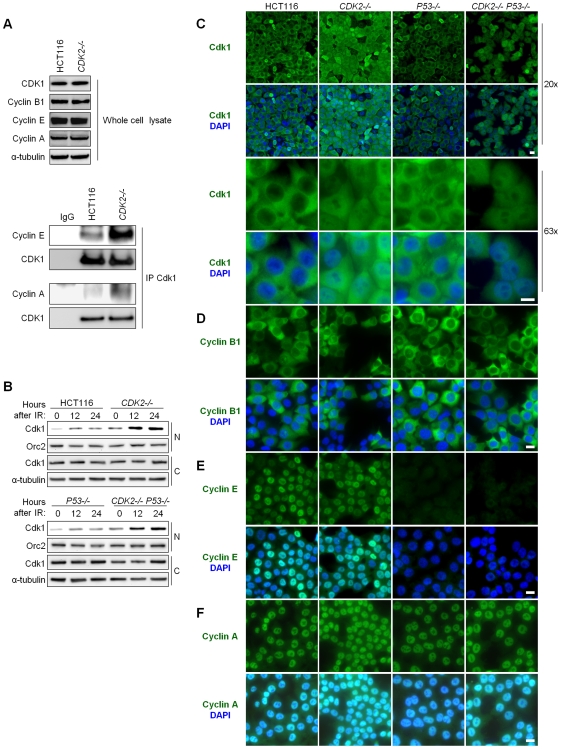
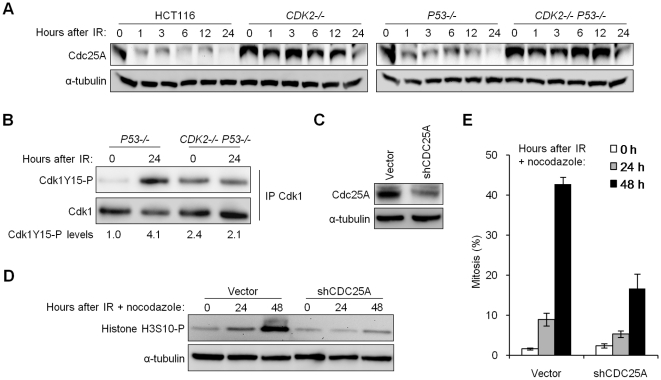
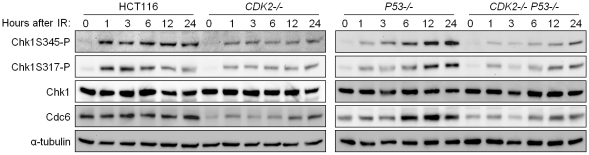
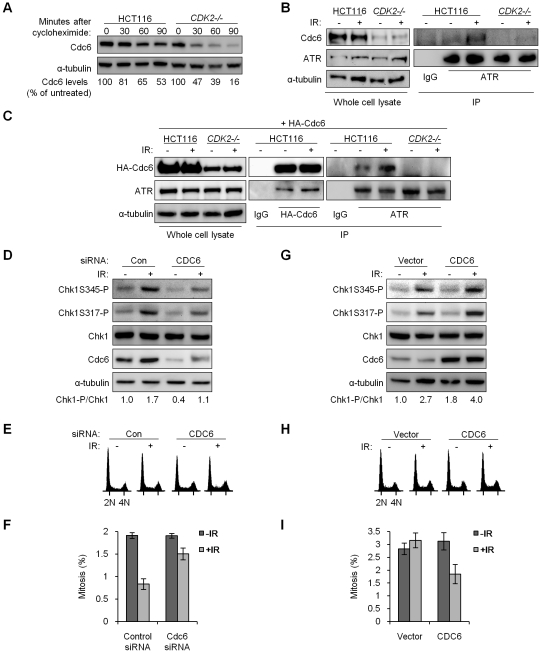
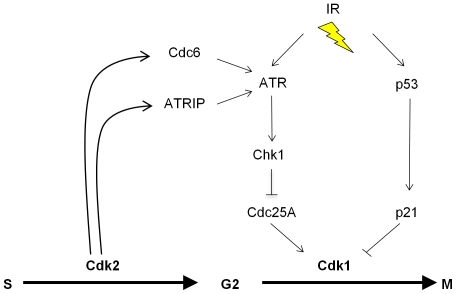
The activation of phase-specific cyclin-dependent kinases (Cdks) is associated with ordered cell cycle transitions. Among the mammalian Cdks, only Cdk1 is essential for somatic cell proliferation. Cdk1 can apparently substitute for Cdk2, Cdk4, and Cdk6, which are individually dispensable in mice. It is unclear if all functions of non-essential Cdks are fully redundant with Cdk1. Using a genetic approach, we show that Cdk2, the S-phase Cdk, uniquely controls the G(2)/M checkpoint that prevents cells with damaged DNA from initiating mitosis. CDK2-nullizygous human cells exposed to ionizing radiation failed to exclude Cdk1 from the nucleus and exhibited a marked defect in G(2)/M arrest that was unmasked by the disruption of P53. The DNA replication licensing protein Cdc6, which is normally stabilized by Cdk2, was physically associated with the checkpoint regulator ATR and was required for efficient ATR-Chk1-Cdc25A signaling. These findings demonstrate that Cdk2 maintains a balance of S-phase regulatory proteins and thereby coordinates subsequent p53-independent G(2)/M checkpoint activation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30(11):630–641. - PubMed
-
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699–2711. - PubMed
-
- Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat Rev Mol Cell Biol. 2008;9(11):910–916. - PubMed
-
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. - PubMed
-
- Berthet C, Kaldis P. Cell-specific responses to loss of cyclin-dependent kinases. Oncogene. 2007;26(31):4469–4477. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
