

Inhibition of DNA methyltransferase induces G2 cell cycle arrest and apoptosis in human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5 signalling
- PMID: 20196786
- PMCID: PMC4516515
- DOI: 10.1111/j.1582-4934.2009.00661.x
Inhibition of DNA methyltransferase induces G2 cell cycle arrest and apoptosis in human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5 signalling
Abstract
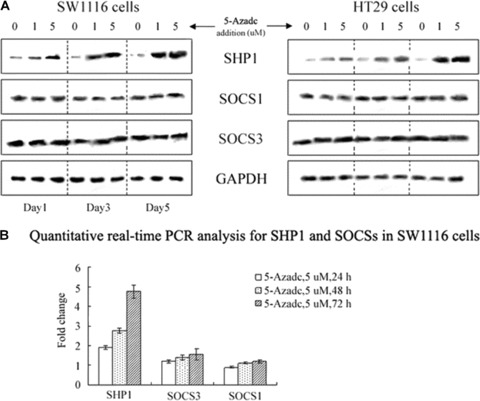
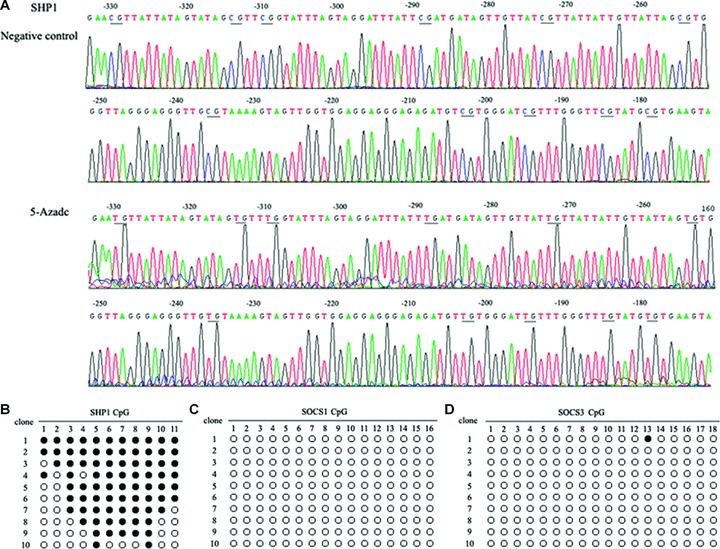
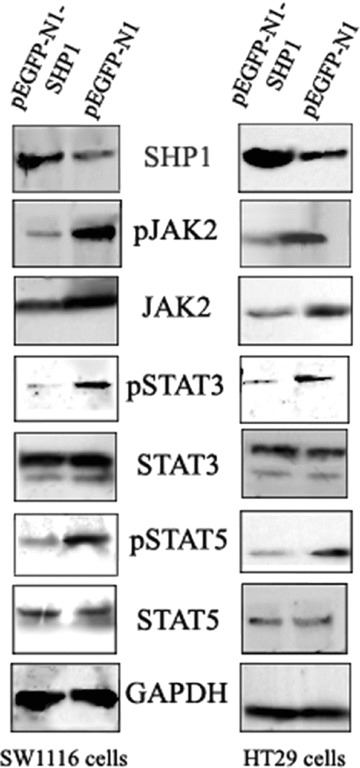
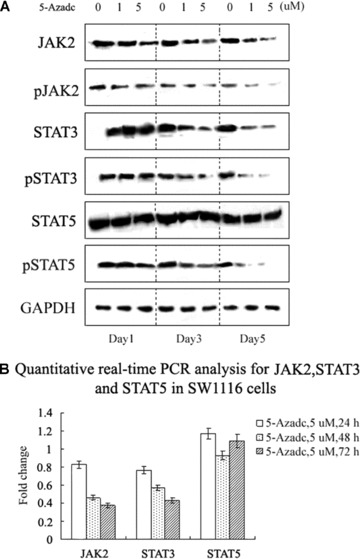
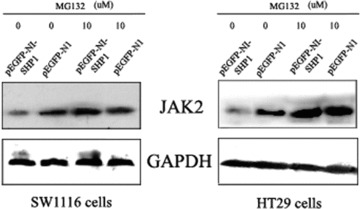
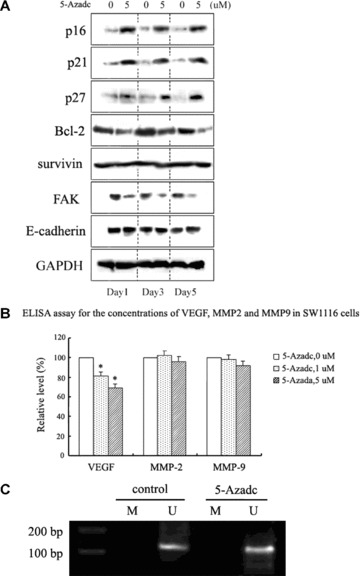
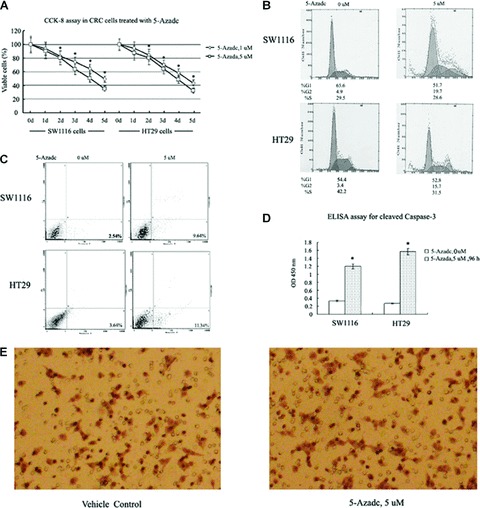
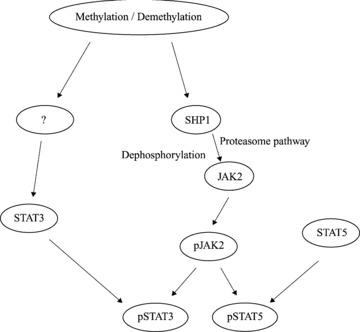
DNA methyltransferase inhibitors (MTIs) have recently emerged as promising chemotherapeutic or preventive agents for cancer, despite their poorly characterized mechanisms of action. The present study shows that DNA methylation is integral to the regulation of SH2-containing protein tyrosine phosphatase 1 (SHP1) expression, but not for regulation of suppressors of cytokine signalling (SOCS)1 or SOCS3 in colorectal cancer (CRC) cells. SHP1 expression correlates with down-regulation of Janus kinase/signal transducers and activators of transcription (JAK2/STAT3/STAT5) signalling, which is mediated in part by tyrosine dephosphorylation events and modulation of the proteasome pathway. Up-regulation of SHP1 expression was achieved using a DNA MTI, 5-aza-2'-deoxycytidine (5-aza-dc), which also generated significant down-regulation of JAK2/STAT3/STAT5 signalling. We demonstrate that 5-aza-dc suppresses growth of CRC cells, and induces G2 cell cycle arrest and apoptosis through regulation of downstream targets of JAK2/STAT3/STAT5 signalling including Bcl-2, p16(ink4a), p21(waf1/cip1) and p27(kip1). Although 5-aza-dc did not significantly inhibit cell invasion, 5-aza-dc did down-regulate expression of focal adhesion kinase and vascular endothelial growth factor in CRC cells. Our results demonstrate that 5-aza-dc can induce SHP1 expression and inhibit JAK2/STAT3/STAT5 signalling. This study represents the first evidence towards establishing a mechanistic link between inhibition of JAK2/STAT3/STAT5 signalling and the anticancer action of 5-aza-dc in CRC cells that may lead to the use of MTIs as a therapeutic intervention for human colorectal cancer.
Figures
References
-
- Lubbert M. DNA methylation inhibitors in the treatment of leukemias, myelodysplastic syndromes and hemoglobinopathies: clinical results and possible mechanisms of action. Curr Top Microbiol Immunol. 2000;249:135–64. - PubMed
-
- Momparler RL, Ayoub J. Potential of 5-aza-2’-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer. 2001;34:S111–5. - PubMed
-
- Issa JP, Kantarjian H. Azacitidine. Nat Rev Drug Discov. 2005:S6–7. - PubMed
-
- Wijermans P, Lubbert M, Verhoef G, et al. Low-dose 5-aza-2’-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–62. - PubMed
-
- Aparicio A, Eads CA, Leong LA, et al. Phase I trial of continuous infusion 5-aza-2′-deoxycytidine. Cancer Chemother Pharmacol. 2003;51:231–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
