

A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Sträussler-Scheinker disease phenotype: comparison with similar cases from the literature
- PMID: 20198483
- PMCID: PMC3015204
- DOI: 10.1007/s00401-010-0656-3
A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Sträussler-Scheinker disease phenotype: comparison with similar cases from the literature
Abstract
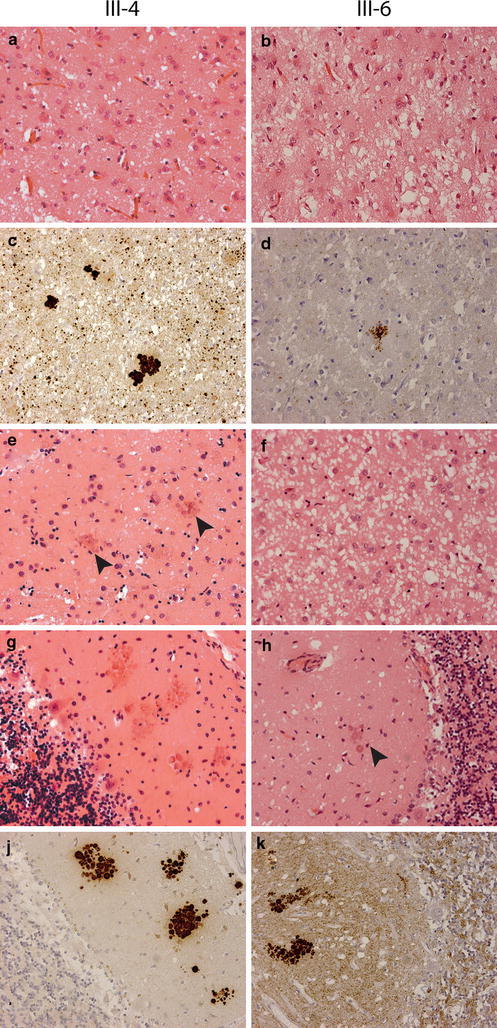
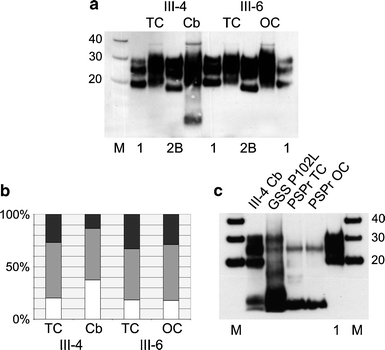
Human prion diseases can be sporadic, inherited or acquired by infection and show considerable phenotypic heterogeneity. We describe the clinical, histopathological and pathological prion protein (PrP(Sc)) characteristics of a Dutch family with a novel 7-octapeptide repeat insertion (7-OPRI) in PRNP, the gene encoding the prion protein (PrP). Clinical features were available in four, neuropathological features in three and biochemical characteristics in two members of this family. The clinical phenotype was characterized by slowly progressive cognitive decline, personality change, lethargy, depression with anxiety and panic attacks, apraxia and a hypokinetic-rigid syndrome. Neuropathological findings consisted of numerous multi- and unicentric amyloid plaques throughout the cerebrum and cerebellum with varying degrees of spongiform degeneration. Genetic and molecular studies were performed in two male family members. One of them was homozygous for valine and the other heterozygous for methionine and valine at codon 129 of PRNP. Sequence analysis identified a novel 168 bp insertion [R2-R2-R2-R2-R3g-R2-R2] in the octapeptide repeat region of PRNP. Both patients carried the mutation on the allele with valine at codon 129. Western blot analysis showed type 1 PrP(Sc) in both patients and detected a smaller ~8 kDa PrP(Sc) fragment in the cerebellum in one patient. The features of this Dutch kindred define an unusual neuropathological phenotype and a novel PRNP haplotype among the previously documented 7-OPRI mutations, further expanding the spectrum of genotype-phenotype correlations in inherited prion diseases.
Figures

References
-
- Brown P, Goldfarb LG, McCombie WR, et al. Atypical Creutzfeldt-Jakob disease in an American family with an insert mutation in the PRNP amyloid precursor gene. Neurology. 1992;42:422–427. - PubMed
-
- Capellari S, Vital C, Parchi P, et al. Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology. 1997;49:133–141. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
