

Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean
- PMID: 20199683
- PMCID: PMC2848761
- DOI: 10.1186/1471-2229-10-41
Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean
Abstract
Background: The nutritional and economic value of many crops is effectively a function of seed protein and oil content. Insight into the genetic and molecular control mechanisms involved in the deposition of these constituents in the developing seed is needed to guide crop improvement. A quantitative trait locus (QTL) on Linkage Group I (LG I) of soybean (Glycine max (L.) Merrill) has a striking effect on seed protein content.
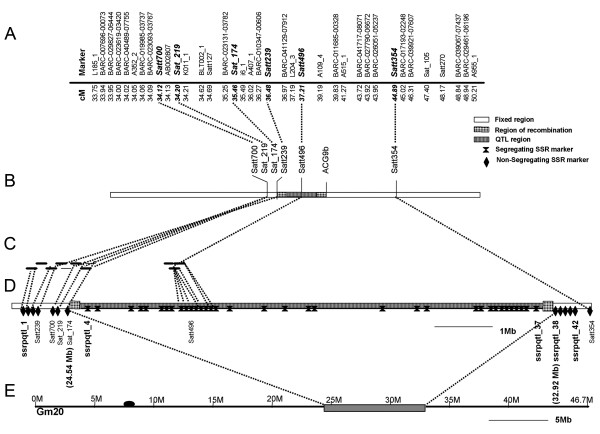
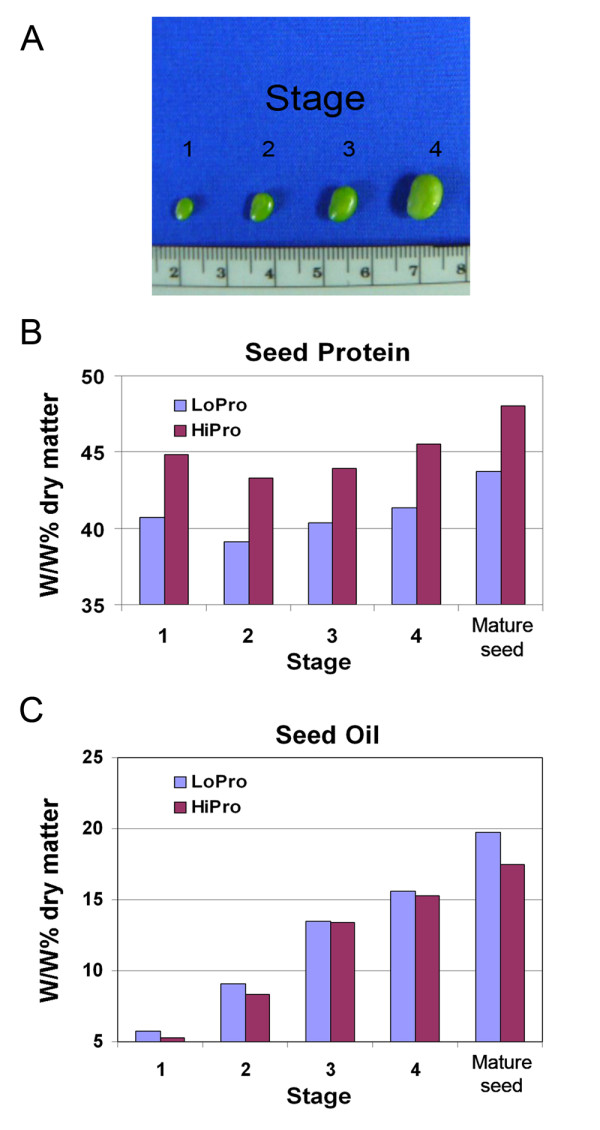
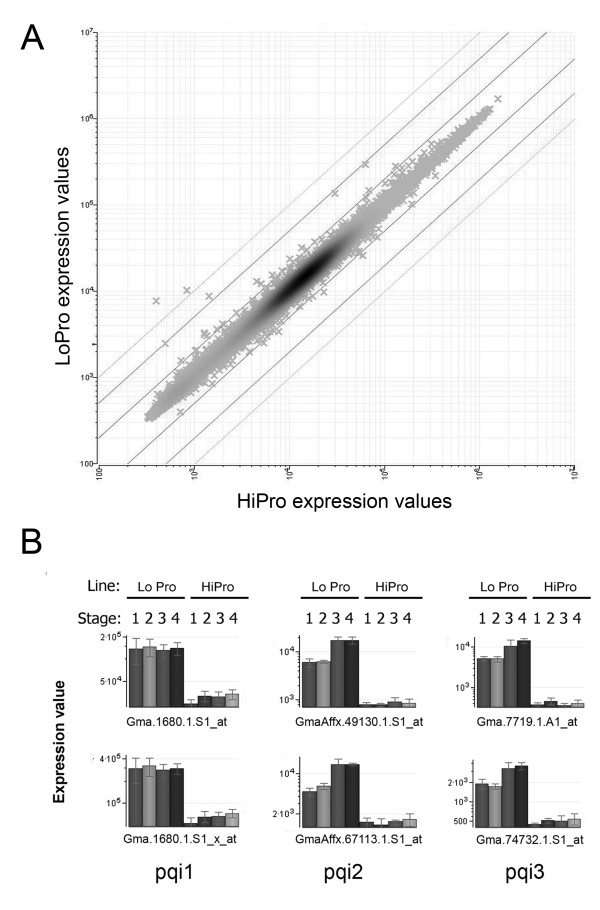
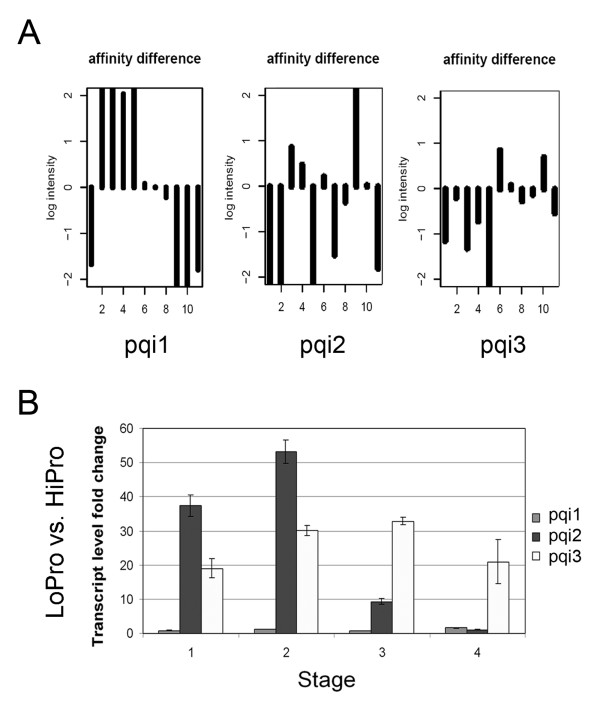
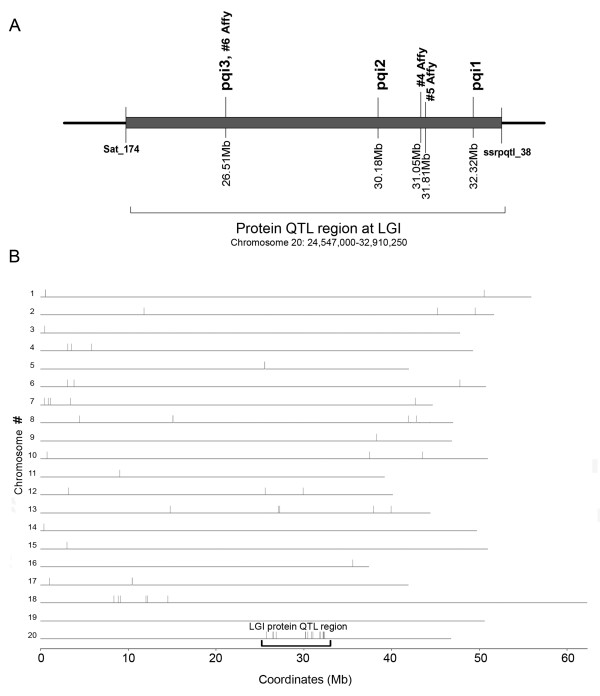
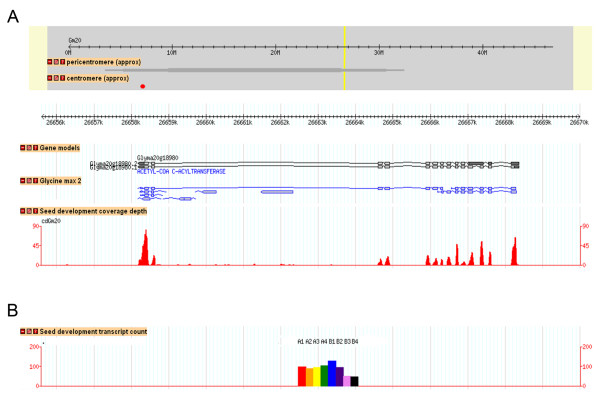
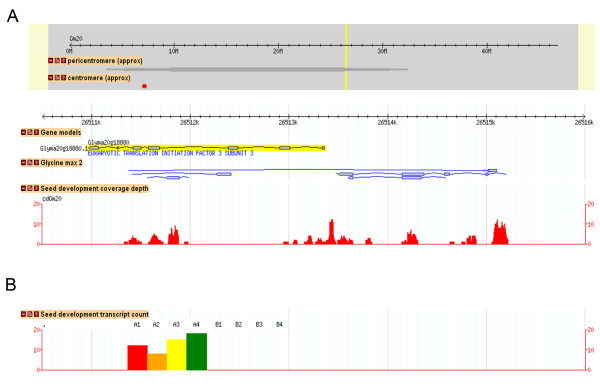
Results: A soybean near-isogenic line (NIL) pair contrasting in seed protein and differing in an introgressed genomic segment containing the LG I protein QTL was used as a resource to demarcate the QTL region and to study variation in transcript abundance in developing seed. The LG I QTL region was delineated to less than 8.4 Mbp of genomic sequence on chromosome 20. Using Affymetrix Soy GeneChip and high-throughput Illumina whole transcriptome sequencing platforms, 13 genes displaying significant seed transcript accumulation differences between NILs were identified that mapped to the 8.4 Mbp LG I protein QTL region.
Conclusions: This study identifies gene candidates at the LG I protein QTL for potential involvement in the regulation of protein content in the soybean seed. The results demonstrate the power of complementary approaches to characterize contrasting NILs and provide genome-wide transcriptome insight towards understanding seed biology and the soybean genome.
Figures
References
-
- Hajduch M, Ganapathy A, Stein JW, Thelen JJ. A systematic proteomic study of seed filling in soybean. Establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol. 2005;137(4):1397–1419. doi: 10.1104/pp.104.056614. - DOI - PMC - PubMed
-
- Gallardo K, Firnhaber C, Zuber H, Hericher D, Belghazi M, Henry C, Kuster H, Thompson R. A combined proteome and transcriptome analysis of developing Medicago truncatula seeds: evidence for metabolic specialization of maternal and filial tissues. Mol Cell Proteomics. 2007;6(12):2165–2179. doi: 10.1074/mcp.M700171-MCP200. - DOI - PubMed
-
- Jukanti AK, Heidlebaugh NM, Parrott DL, Fisher IA, McInnerney K, Fischer AM. Comparative transcriptome profiling of near-isogenic barley (Hordeum vulgare) lines differing in the allelic state of a major grain protein content locus identifies genes with possible roles in leaf senescence and nitrogen reallocation. New Phytol. 2008;177:333–349. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
