

Acute kidney injury: a springboard for progression in chronic kidney disease
- PMID: 20200097
- PMCID: PMC2867413
- DOI: 10.1152/ajprenal.00017.2010
Acute kidney injury: a springboard for progression in chronic kidney disease
Abstract
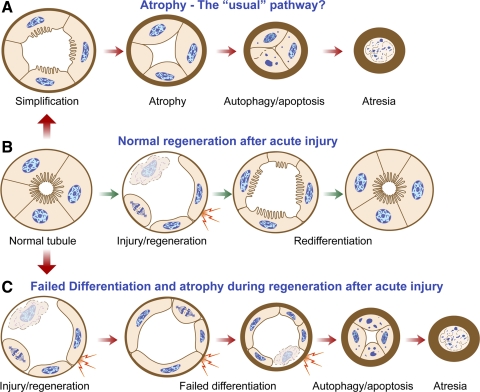
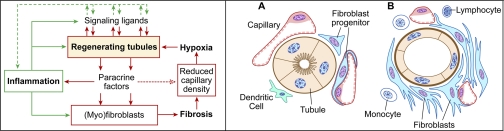
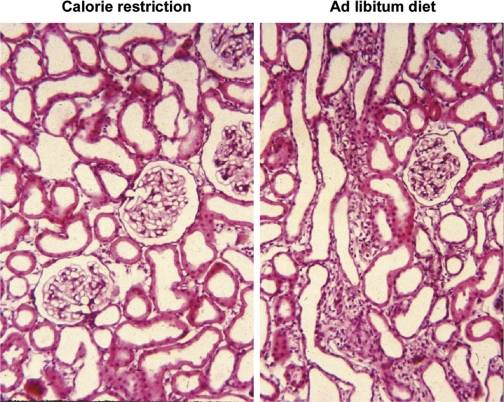
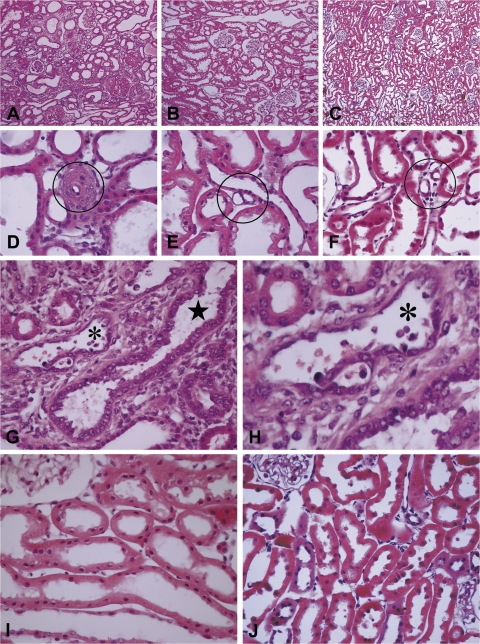
Recently published epidemiological and outcome analysis studies have brought to our attention the important role played by acute kidney injury (AKI) in the progression of chronic kidney disease (CKD) to end-stage renal disease (ESRD). AKI accelerates progression in patients with CKD; conversely, CKD predisposes patients to AKI. This research gives credence to older, well-thought-out wisdom that recovery from AKI is often not complete and is marked by residual structural damage. It also mirrors older experimental observations showing that unilateral nephrectomy, a surrogate for loss of nephrons by disease, compromises structural recovery and worsens tubulointerstitial fibrosis after ischemic AKI. Moreover, review of a substantial body of work on the relationships among reduced renal mass, hypertension, and pathology associated with these conditions suggests that impaired myogenic autoregulation of blood flow in the setting of hypertension, the arteriolosclerosis that results, and associated recurrent ischemic AKI in microscopic foci play important roles in the development of progressively increasing tubulointerstitial fibrosis. How nutrition, an additional factor that profoundly affects renal disease progression, influences these events needs reevaluation in light of information on the effects of calories vs. protein and animal vs. vegetable protein on injury and progression. Considerations based on published and emerging data suggest that a pathology that develops in regenerating tubules after AKI characterized by failure of differentiation and persistently high signaling activity is the proximate cause that drives downstream events in the interstitium: inflammation, capillary rarefaction, and fibroblast proliferation. In light of this information, we advance a comprehensive hypothesis regarding the pathophysiology of AKI as it relates to the progression of kidney disease. We discuss the implications of this pathophysiology for developing efficient therapeutic strategies to delay progression and avert ESRD.
Figures
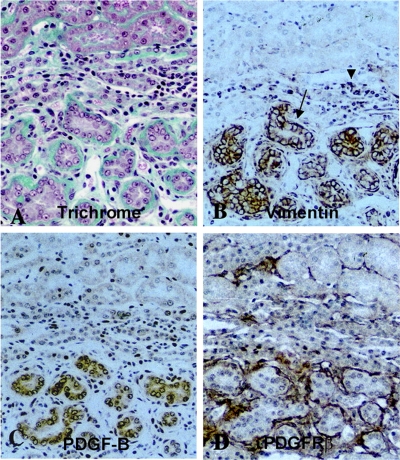
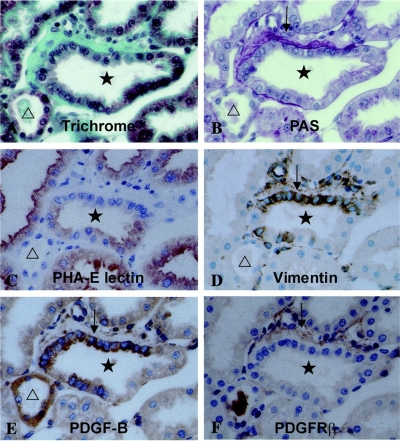
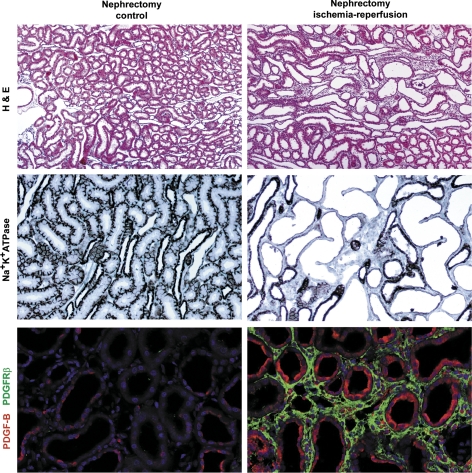
References
-
- Abrams JR, Tapp DC, Venkatachalam MA. Dietary influence and pathologic changes. In: Progressive Nature of Renal Disease, edited by Mitch WE. New York: Churchill Livingstone, 1992, p. 133–148
-
- Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol 24: 46–53, 2004 - PubMed
-
- Abrass CK. Overview: obesity: what does it have to do with kidney disease? J Am Soc Nephrol 15: 2768–2772, 2004 - PubMed
-
- Abu-Amarah I, Bidani AK, Hacioglu R, Williamson GA, Griffin KA. Differential effects of salt on renal hemodynamics and potential pressure transmission in stroke-prone and stroke-resistant spontaneously hypertensive rats. Am J Physiol Renal Physiol 289: F305–F313, 2005 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical