

Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis
- PMID: 20200273
- PMCID: PMC2865890
- DOI: 10.4049/jimmunol.0903191
Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis
Abstract
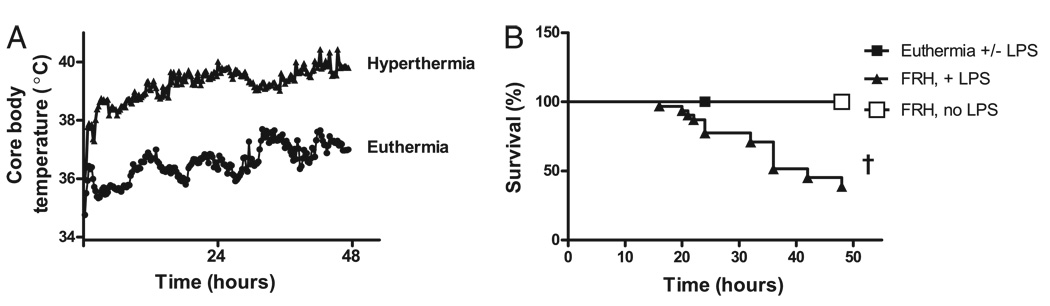
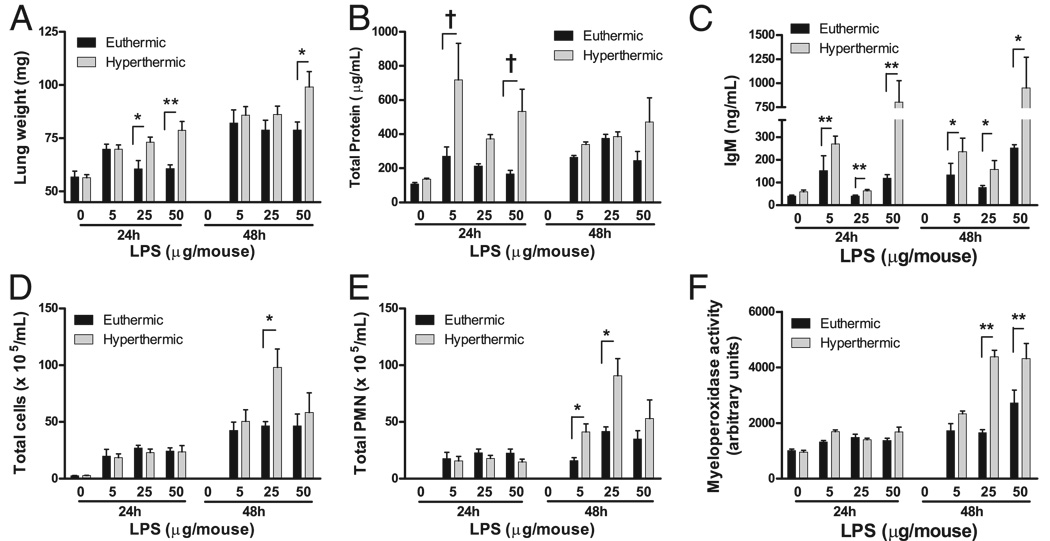
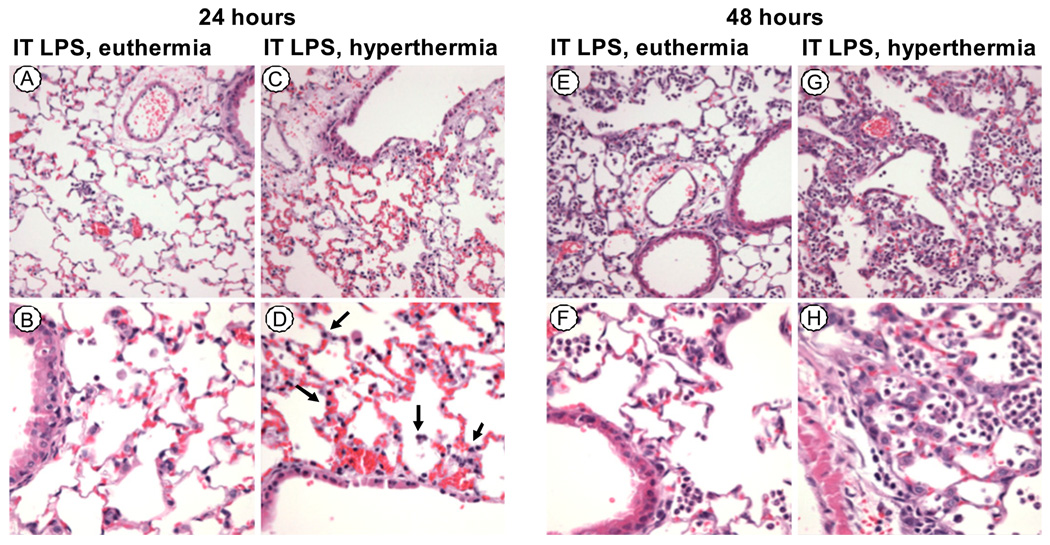
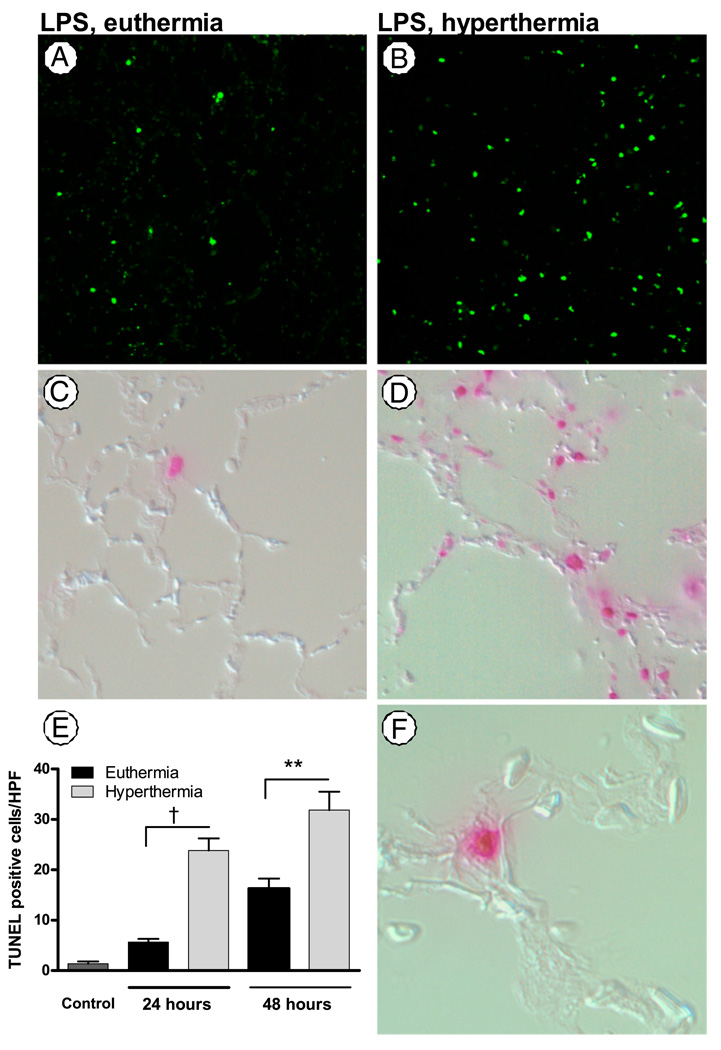
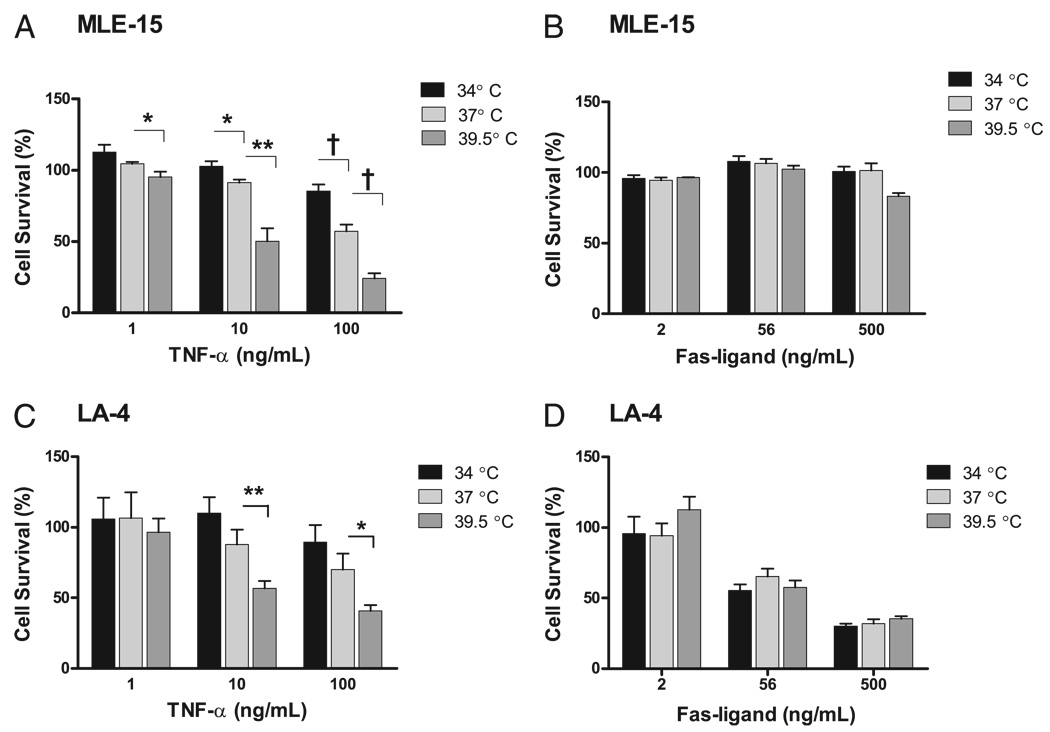
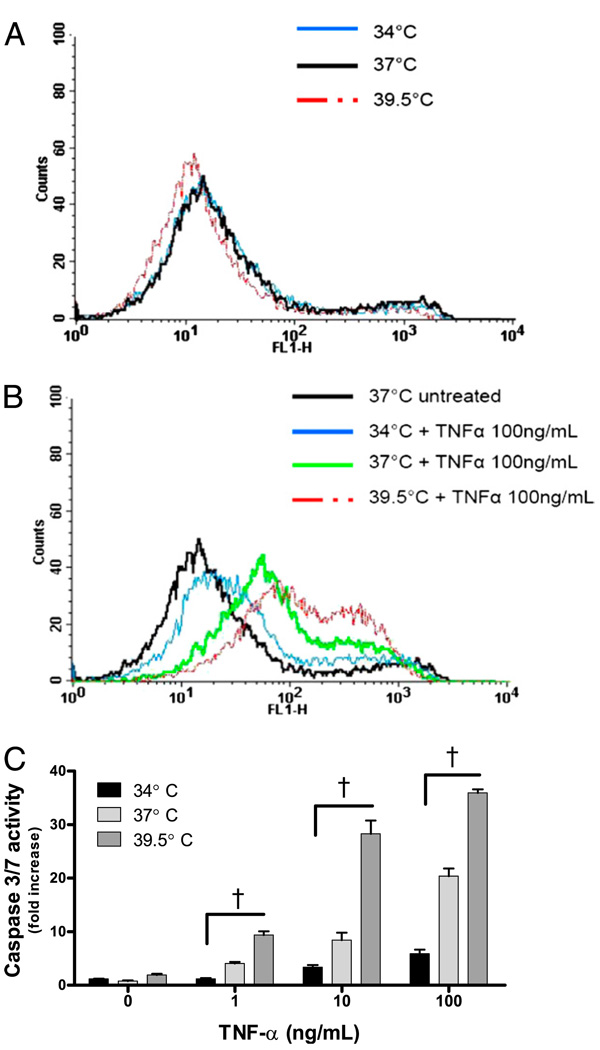
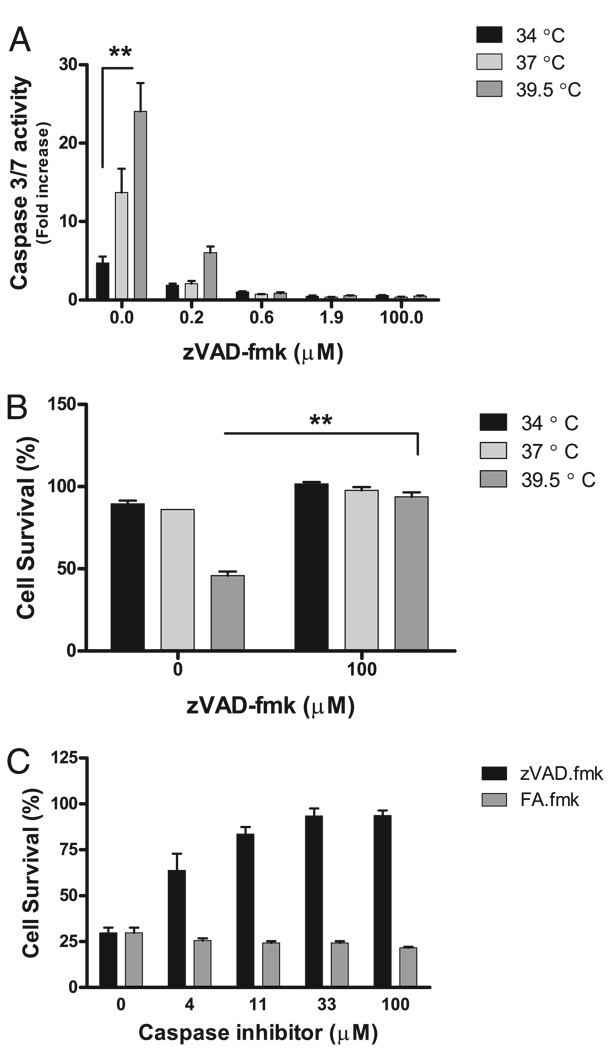
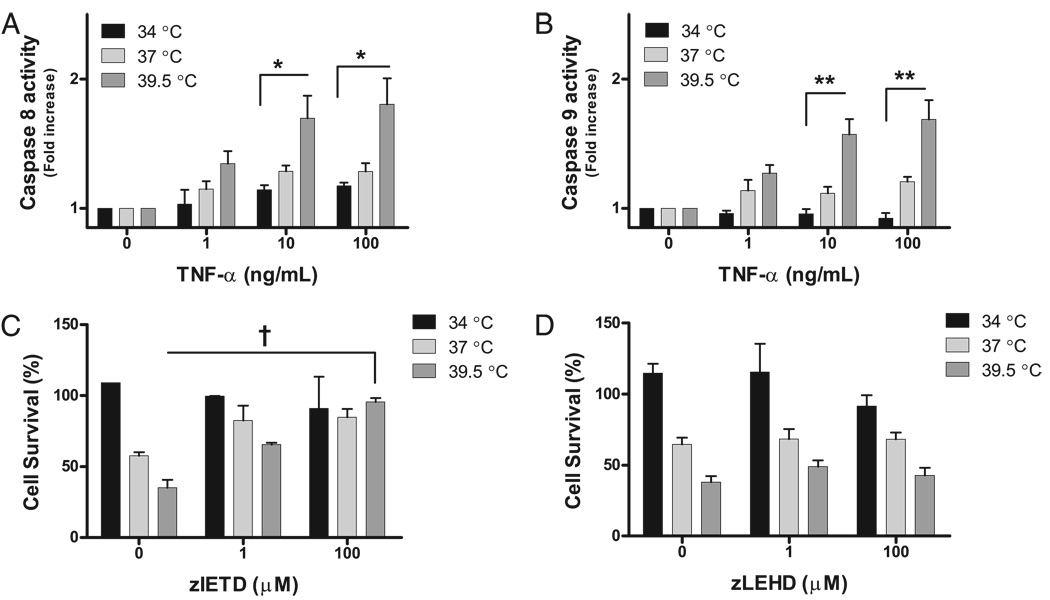
Fever is common in critically ill patients and is associated with worse clinical outcomes, including increased intensive care unit mortality. In animal models, febrile-range hyperthermia (FRH) worsens acute lung injury, but the mechanisms by which this occurs remain uncertain. We hypothesized that FRH augments the response of the alveolar epithelium to TNF-alpha receptor family signaling. We found that FRH augmented LPS-induced lung injury and increased LPS-induced mortality in mice. At 24 h, animals exposed to hyperthermia and LPS had significant increases in alveolar permeability without changes in inflammatory cells in bronchoalveolar lavage fluid or lung tissue as compared with animals exposed to LPS alone. The increase in alveolar permeability was associated with an increase in alveolar epithelial apoptosis and was attenuated by caspase inhibition with zVAD.fmk. At 48 h, the animals exposed to hyperthermia and LPS had an enhanced lung inflammatory response. In murine lung epithelial cell lines (MLE-15, LA-4) and in primary type II alveolar epithelial cells, FRH enhanced apoptosis in response to TNF-alpha but not Fas ligand. The increase in apoptosis was caspase-8 dependent and associated with suppression of NF-kappaB activity. The FRH-associated NF-kappaB suppression was not associated with persistence of IkappaB-alpha, suggesting that FRH-mediated suppression of NF-kappaB occurs by means other than alteration of IkappaB-alpha kinetics. These data show for the first time that FRH promotes lung injury in part by increasing lung epithelial apoptosis. The enhanced apoptotic response might relate to FRH-mediated suppression of NF-kappaB activity in the alveolar epithelium with a resultant increase in susceptibility to TNF-alpha-mediated cell death.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
Similar articles
-
Death receptors mediate the adverse effects of febrile-range hyperthermia on the outcome of lipopolysaccharide-induced lung injury.Am J Physiol Lung Cell Mol Physiol. 2011 Jul;301(1):L60-70. doi: 10.1152/ajplung.00314.2010. Epub 2011 Apr 22. Am J Physiol Lung Cell Mol Physiol. 2011. PMID: 21515659 Free PMC article.
-
Febrile-range hyperthermia augments neutrophil accumulation and enhances lung injury in experimental gram-negative bacterial pneumonia.J Immunol. 2005 Mar 15;174(6):3676-85. doi: 10.4049/jimmunol.174.6.3676. J Immunol. 2005. PMID: 15749906
-
Inhibition of Acute Lung Injury by TNFR-Fc through Regulation of an Inflammation-Oxidative Stress Pathway.PLoS One. 2016 Mar 18;11(3):e0151672. doi: 10.1371/journal.pone.0151672. eCollection 2016. PLoS One. 2016. PMID: 26990441 Free PMC article.
-
Inhibition of mammalian target of rapamycin augments lipopolysaccharide-induced lung injury and apoptosis.J Immunol. 2012 May 1;188(9):4535-42. doi: 10.4049/jimmunol.1003655. Epub 2012 Mar 26. J Immunol. 2012. PMID: 22450807
-
Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury.Am J Respir Crit Care Med. 2008 Aug 15;178(4):356-62. doi: 10.1164/rccm.200707-1069OC. Epub 2008 Jun 5. Am J Respir Crit Care Med. 2008. PMID: 18535257
Cited by
-
Activation of heat shock response augments fibroblast growth factor-1 expression in wounded lung epithelium.Am J Physiol Lung Cell Mol Physiol. 2016 Nov 1;311(5):L941-L955. doi: 10.1152/ajplung.00262.2016. Epub 2016 Sep 16. Am J Physiol Lung Cell Mol Physiol. 2016. PMID: 27638903 Free PMC article.
-
Primed Mesenchymal Stem Cells by IFN-γ and IL-1β Ameliorate Acute Respiratory Distress Syndrome through Enhancing Homing Effect and Immunomodulation.Biomol Ther (Seoul). 2025 Mar 1;33(2):311-324. doi: 10.4062/biomolther.2025.004. Epub 2025 Feb 20. Biomol Ther (Seoul). 2025. PMID: 39973472 Free PMC article.
-
Shifts in temperature within the physiologic range modify strand-specific expression of select human microRNAs.RNA. 2015 Jul;21(7):1261-73. doi: 10.1261/rna.049122.114. Epub 2015 May 27. RNA. 2015. PMID: 26018549 Free PMC article.
-
Fever integrates antimicrobial defences, inflammation control, and tissue repair in a cold-blooded vertebrate.Elife. 2023 Mar 14;12:e83644. doi: 10.7554/eLife.83644. Elife. 2023. PMID: 36917159 Free PMC article.
-
New insights into the mechanisms of pulmonary edema in acute lung injury.Ann Transl Med. 2018 Jan;6(2):32. doi: 10.21037/atm.2017.12.18. Ann Transl Med. 2018. PMID: 29430449 Free PMC article. Review.
References
-
- Laupland KB, Shahpori R, Kirkpatrick AW, Ross T, Gregson DB, Stelfox HT. Occurrence and outcome of fever in critically ill adults. Crit. Care Med. 2008;36:1531–1535. - PubMed
-
- Peres Bota D, Lopes Ferreira F, Mélot C, Vincent JL. Body temperature alterations in the critically ill. Intensive Care Med. 2004;30:811–816. - PubMed
-
- Circiumaru B, Baldock G, Cohen J. A prospective study of fever in the intensive care unit. Intensive Care Med. 1999;25:668–673. - PubMed
-
- Swenson BR, Hedrick TL, Popovsky K, Pruett TL, Sawyer RG. Is fever protective in surgical patients with bloodstream infection? J. Am. Coll. Surg. 2007;204:815–821. discussion 822-813. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
