

Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice
- PMID: 20200449
- PMCID: PMC2846040
- DOI: 10.1172/JCI39492
Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice
Abstract
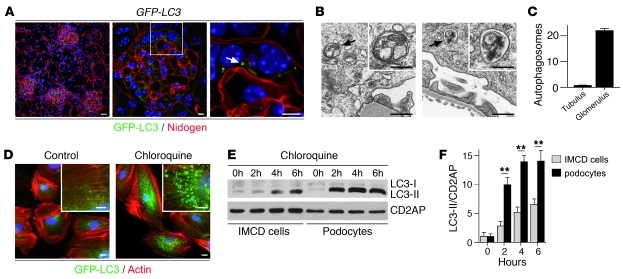
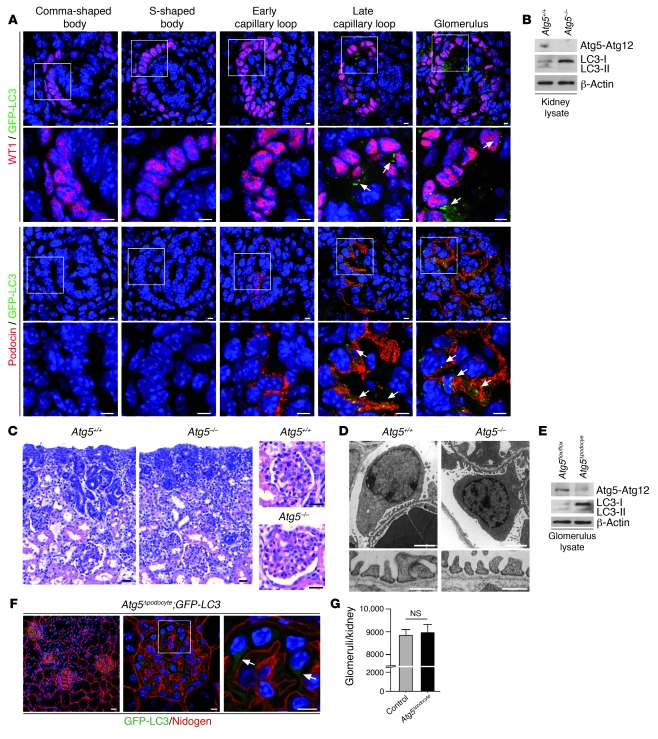
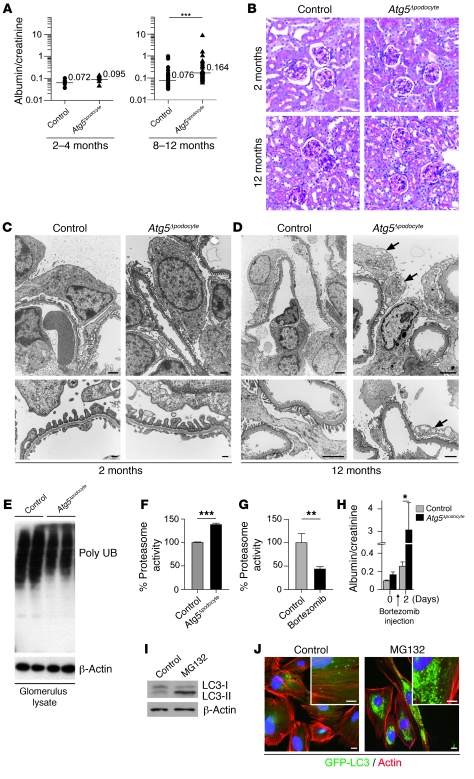
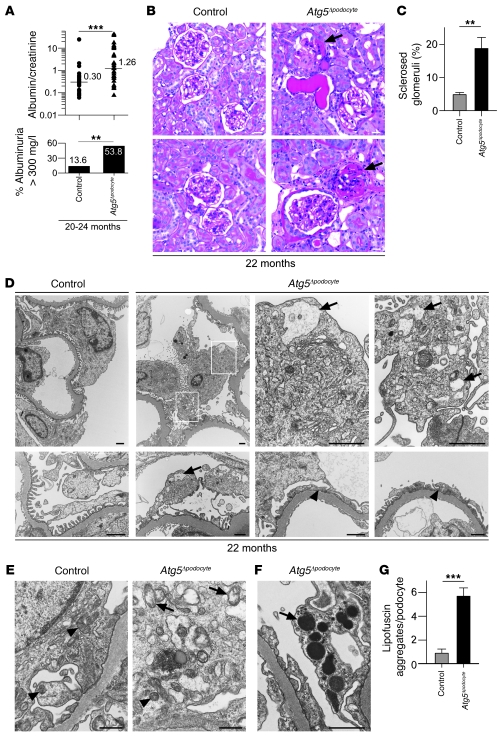
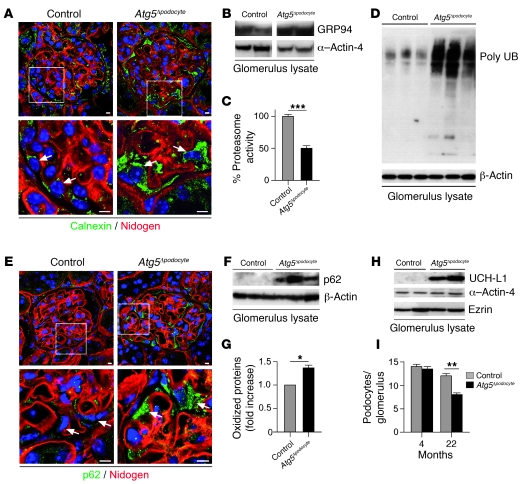
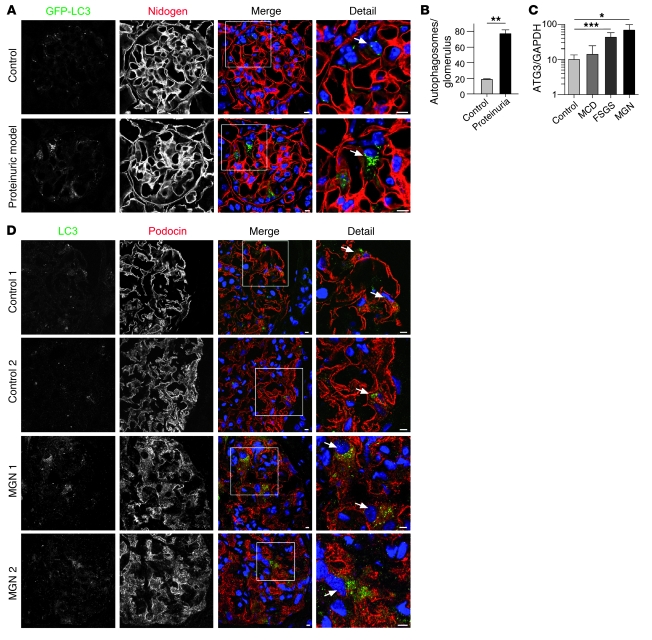
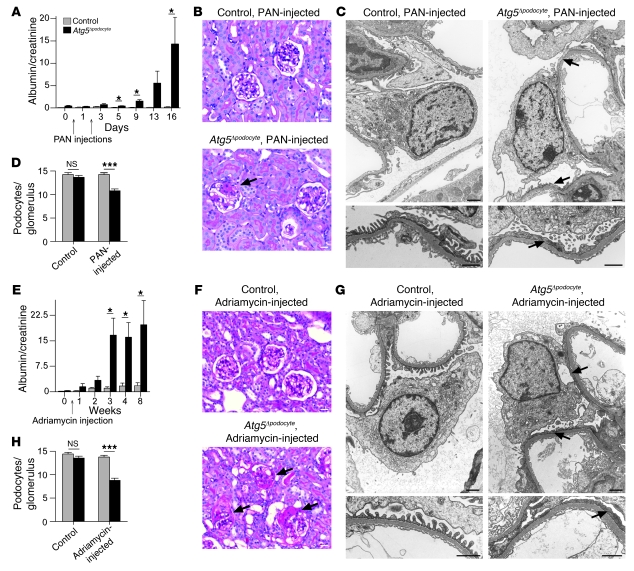
Injury and loss of podocytes are leading factors of glomerular disease and renal failure. The postmitotic podocyte is the primary glomerular target for toxic, immune, metabolic, and oxidant stress, but little is known about how this cell type copes with stress. Recently, autophagy has been identified as a major pathway that delivers damaged proteins and organelles to lysosomes in order to maintain cellular homeostasis. Here we report that podocytes exhibit an unusually high level of constitutive autophagy. Podocyte-specific deletion of autophagy-related 5 (Atg5) led to a glomerulopathy in aging mice that was accompanied by an accumulation of oxidized and ubiquitinated proteins, ER stress, and proteinuria. These changes resulted ultimately in podocyte loss and late-onset glomerulosclerosis. Analysis of pathophysiological conditions indicated that autophagy was substantially increased in glomeruli from mice with induced proteinuria and in glomeruli from patients with acquired proteinuric diseases. Further, mice lacking Atg5 in podocytes exhibited strongly increased susceptibility to models of glomerular disease. These findings highlight the importance of induced autophagy as a key homeostatic mechanism to maintain podocyte integrity. We postulate that constitutive and induced autophagy is a major protective mechanism against podocyte aging and glomerular injury, representing a putative target to ameliorate human glomerular disease and aging-related loss of renal function.
Figures
References
-
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83(1):253–307. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
