

Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance
- PMID: 20200450
- PMCID: PMC2846044
- DOI: 10.1172/JCI39987
Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance
Abstract
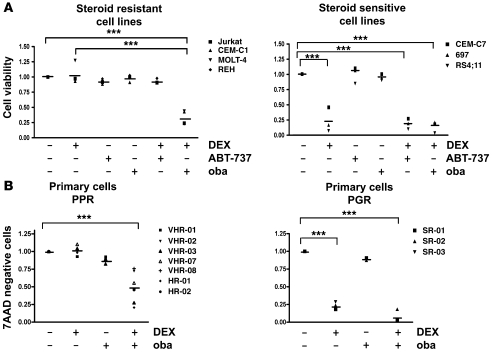
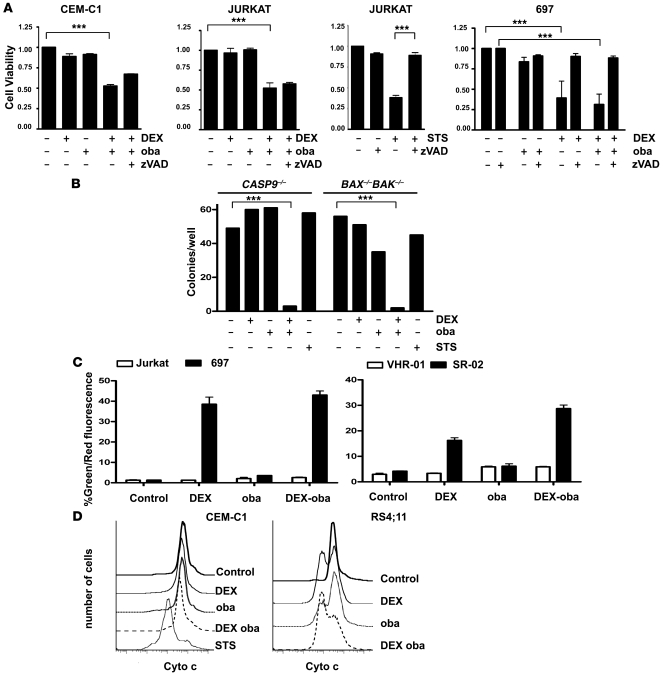
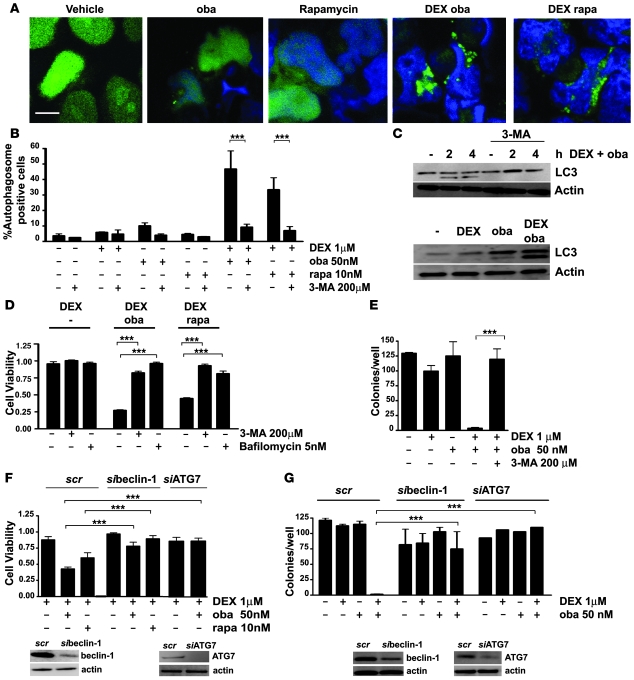
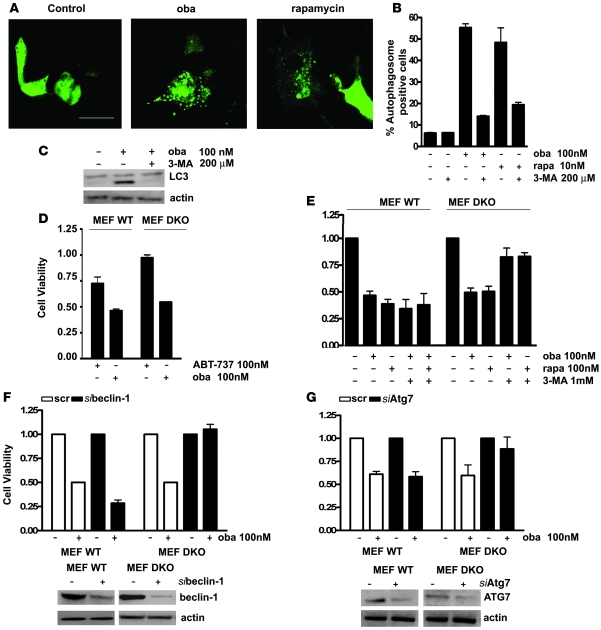
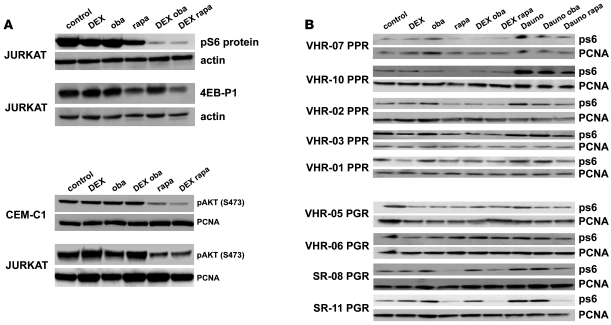
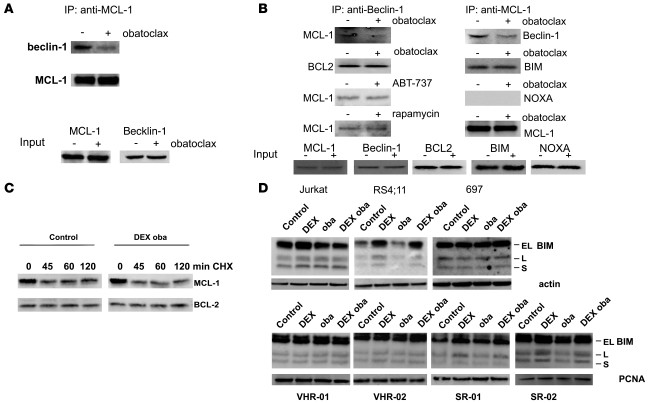
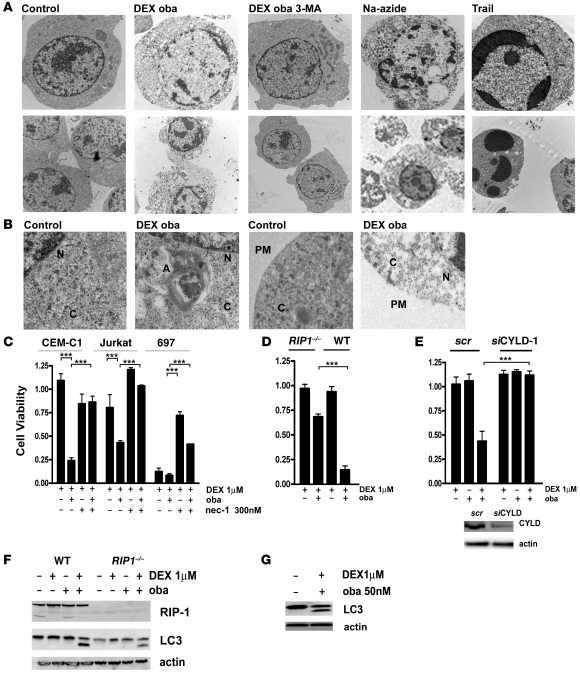
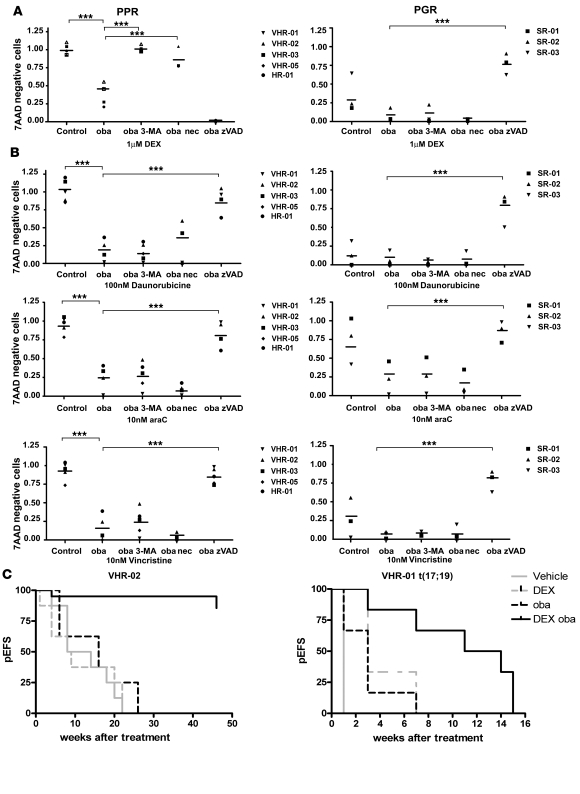
In vivo resistance to first-line chemotherapy, including to glucocorticoids, is a strong predictor of poor outcome in children with acute lymphoblastic leukemia (ALL). Modulation of cell death regulators represents an attractive strategy for subverting such drug resistance. Here we report complete resensitization of multidrug-resistant childhood ALL cells to glucocorticoids and other cytotoxic agents with subcytotoxic concentrations of obatoclax, a putative antagonist of BCL-2 family members. The reversal of glucocorticoid resistance occurred through rapid activation of autophagy-dependent necroptosis, which bypassed the block in mitochondrial apoptosis. This effect was associated with dissociation of the autophagy inducer beclin-1 from the antiapoptotic BCL-2 family member myeloid cell leukemia sequence 1 (MCL-1) and with a marked decrease in mammalian target of rapamycin (mTOR) activity. Consistent with a protective role for mTOR in glucocorticoid resistance in childhood ALL, combination of rapamycin with the glucocorticoid dexamethasone triggered autophagy-dependent cell death, with characteristic features of necroptosis. Execution of cell death, but not induction of autophagy, was strictly dependent on expression of receptor-interacting protein (RIP-1) kinase and cylindromatosis (turban tumor syndrome) (CYLD), two key regulators of necroptosis. Accordingly, both inhibition of RIP-1 and interference with CYLD restored glucocorticoid resistance completely. Together with evidence for a chemosensitizing activity of obatoclax in vivo, our data provide a compelling rationale for clinical translation of this pharmacological approach into treatments for patients with refractory ALL.
Figures
References
-
- Schrappe M. Evolution of BFM trials for childhood ALL. Ann Hematol. 2004;83(Suppl 1):S121–S123. - PubMed
-
- Schrauder A, et al. Prospective evaluation of MRD–Kinetics in 274 children with high-risk ALL treated in trial ALL–BFM 2000: Insights into development of resistance and impact on further refinement of treatment stratification strategies. Blood. 2007;110:585.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
