

Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures
- PMID: 20201486
- PMCID: PMC2948786
- DOI: 10.1021/ar900165x
Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures
Abstract
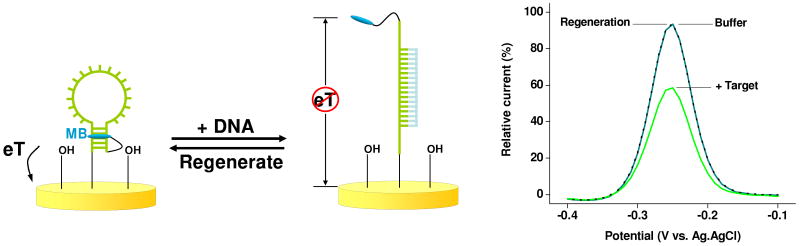
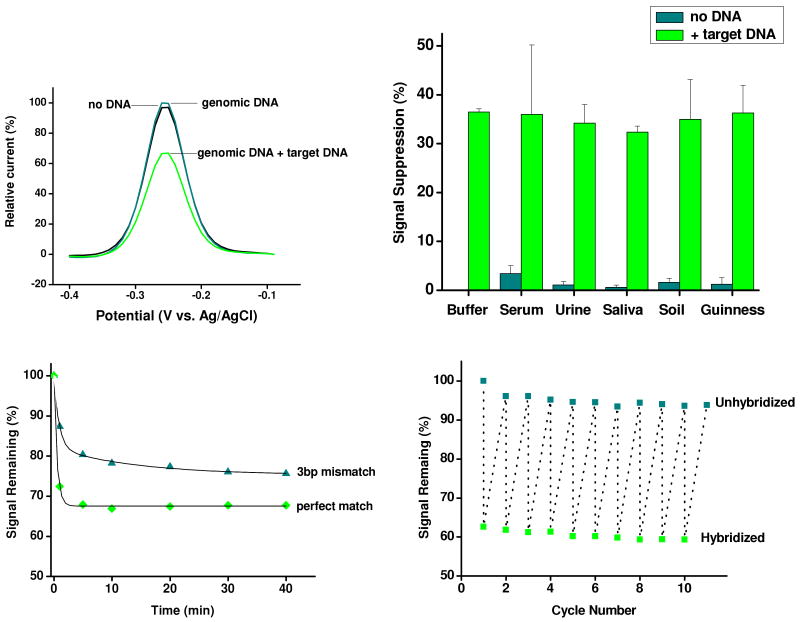
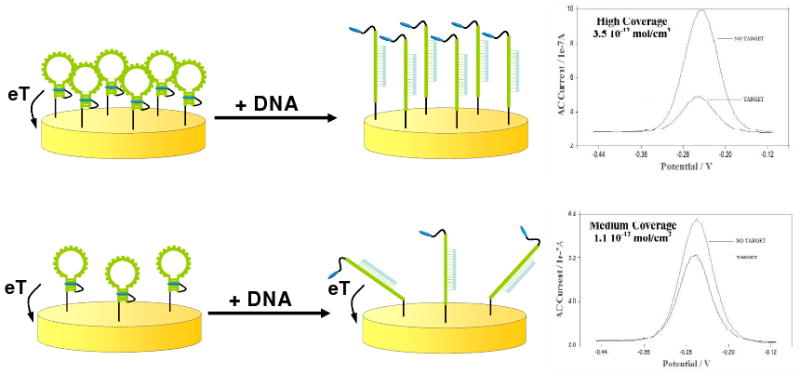
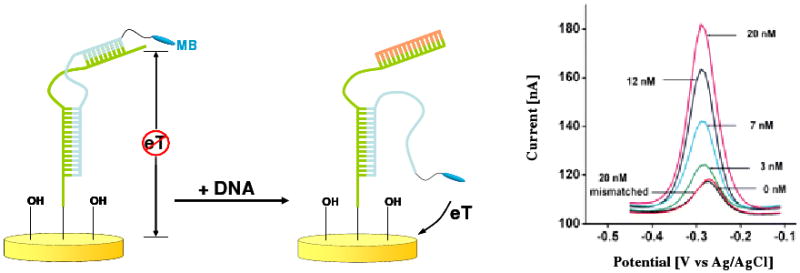
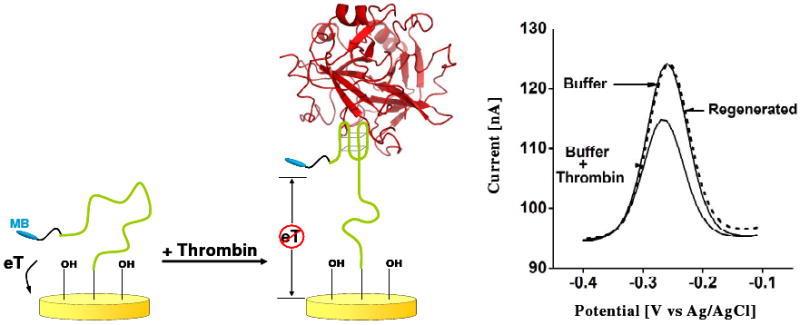
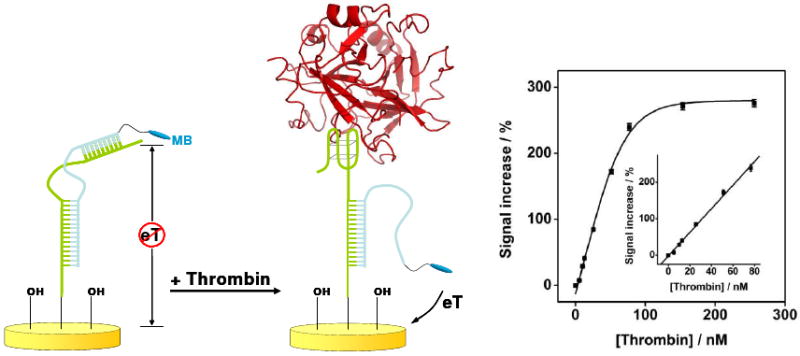
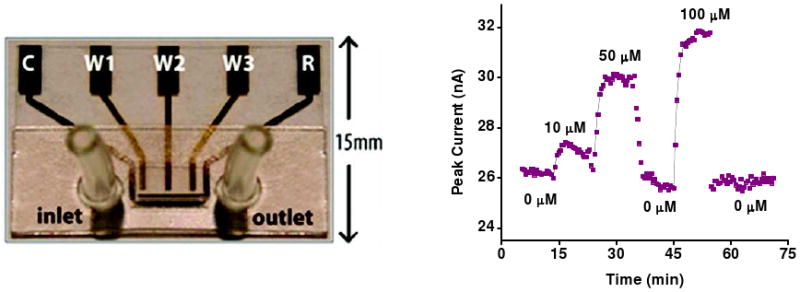
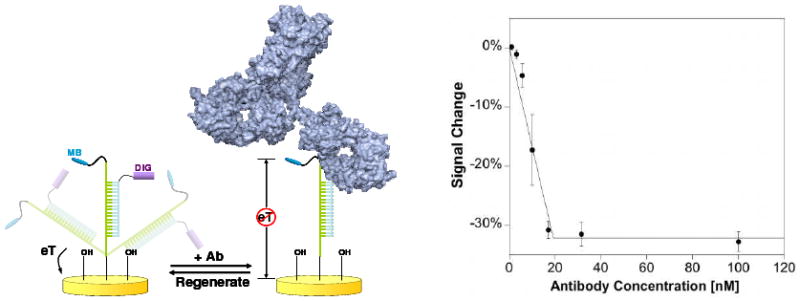
Biomolecular recognition is versatile, specific, and high affinity, qualities that have motivated decades of research aimed at adapting biomolecules into a general platform for molecular sensing. Despite significant effort, however, so-called "biosensors" have almost entirely failed to achieve their potential as reagentless, real-time analytical devices; the only quantitative, reagentless biosensor to achieve commercial success so far is the home glucose monitor, employed by millions of diabetics. The fundamental stumbling block that has precluded more widespread success of biosensors is the failure of most biomolecules to produce an easily measured signal upon target binding. Antibodies, for example, do not change their shape or dynamics when they bind their recognition partners, nor do they emit light or electrons upon binding. It has thus proven difficult to transduce biomolecular binding events into a measurable output signal, particularly one that is not readily spoofed by the binding of any of the many potentially interfering species in typical biological samples. Analytical approaches based on biomolecular recognition are therefore mostly cumbersome, multistep processes relying on analyte separation and isolation (such as Western blots, ELISA, and other immunochemical methods); these techniques have proven enormously useful, but are limited almost exclusively to laboratory settings. In this Account, we describe how we have refined a potentially general solution to the problem of signal detection in biosensors, one that is based on the binding-induced "folding" of electrode-bound DNA probes. That is, we have developed a broad new class of biosensors that employ electrochemistry to monitor binding-induced changes in the rigidity of a redox-tagged probe DNA that has been site-specifically attached to an interrogating electrode. These folding-based sensors, which have been generalized to a wide range of specific protein, nucleic acid, and small-molecule targets, are rapid (responding in seconds to minutes), sensitive (detecting sub-picomolar to micromolar concentrations), and reagentless. They are also greater than 99% reusable, are supported on micrometer-scale electrodes, and are readily fabricated into densely packed sensor arrays. Finally, and critically, their signaling is linked to a binding-specific change in the physics of the probe DNA, and not simply to adsorption of the target onto the sensor head. Accordingly, they are selective enough to be employed directly in blood, crude soil extracts, cell lysates, and other grossly contaminated clinical and environmental samples. Indeed, we have recently demonstrated the ability to quantitatively monitor a specific small molecule in real-time directly in microliters of flowing, unmodified blood serum. Because of their sensitivity, substantial background suppression, and operational convenience, these folding-based biosensors appear potentially well suited for electronic, on-chip applications in pathogen detection, proteomics, metabolomics, and drug discovery.
Figures
References
-
- Engvall E, Perlman P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. - PubMed
-
- Van Weemen BK, Schuurs AH. Immunoassay using antigen-enzyme conjugates. FEBS Letters. 1971;15:232–236. - PubMed
-
- Clark LC, Lyons C. Electrode Systems for Continuous Monitoring in Cardiovascular Surgery. Ann NY Acad Sci. 1962;102:29–45. - PubMed
-
- Updike SJ, Hicks GP. The enzyme electrode. Nature. 1967;214:986–988. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
