

Review: autophagy and neurodegeneration: survival at a cost?
- PMID: 20202120
- PMCID: PMC2860012
- DOI: 10.1111/j.1365-2990.2010.01062.x
Review: autophagy and neurodegeneration: survival at a cost?
Abstract
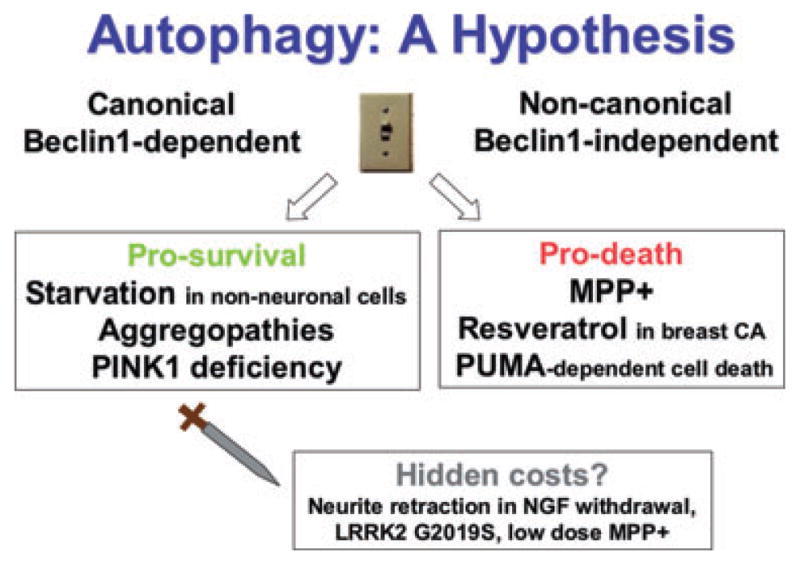
Protein aggregation, mitochondrial impairment and oxidative stress are common to multiple neurodegenerative diseases. Homeostasis is regulated by a balanced set of anabolic and catabolic responses, which govern removal and repair of damaged proteins and organelles. Macroautophagy is an evolutionarily conserved pathway for the degradation of long-lived proteins, effete organelles and protein aggregates. Aberrations in macroautophagy have been observed in Alzheimer, Huntington, Parkinson, motor neuron and prion diseases. In this review, we will discuss the divergent roles of macroautophagy in neurodegenerative diseases and suggest a potential regulatory mechanism that could determine cell death or survival outcomes. We also highlight emerging data on neurite morphology and synaptic remodelling that indicate the possibility of detrimental functional trade-offs in the face of neuronal cell survival, particularly if the need for elevated macroautophagy is sustained.
Figures


Similar articles
-
Targeting autophagy in the brain: a promising approach?Cent Nerv Syst Agents Med Chem. 2010 Jun 1;10(2):158-68. doi: 10.2174/187152410791196350. Cent Nerv Syst Agents Med Chem. 2010. PMID: 20518730 Review.
-
Dining in with BCL-2: new guests at the autophagy table.Clin Sci (Lond). 2009 Oct 26;118(3):173-81. doi: 10.1042/CS20090310. Clin Sci (Lond). 2009. PMID: 19845510 Review.
-
Autophagy in the central nervous system: implications for neurodegenerative disorders.CNS Neurol Disord Drug Targets. 2010 Dec;9(6):701-19. doi: 10.2174/187152710793237421. CNS Neurol Disord Drug Targets. 2010. PMID: 20942791 Review.
-
Autophagy and its normal and pathogenic states in the brain.Annu Rev Neurosci. 2014;37:55-78. doi: 10.1146/annurev-neuro-071013-014149. Epub 2014 Apr 21. Annu Rev Neurosci. 2014. PMID: 24821313 Review.
-
Neuronal autophagy and neurodegenerative diseases.Exp Mol Med. 2012 Feb 29;44(2):89-98. doi: 10.3858/emm.2012.44.2.031. Exp Mol Med. 2012. PMID: 22257884 Free PMC article. Review.
Cited by
-
The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease.Cold Spring Harb Perspect Med. 2012 Aug 1;2(8):a006361. doi: 10.1101/cshperspect.a006361. Cold Spring Harb Perspect Med. 2012. PMID: 22908190 Free PMC article.
-
Potential mechanisms underlying lithium treatment for Alzheimer's disease and COVID-19.Eur Rev Med Pharmacol Sci. 2022 Mar;26(6):2201-2214. doi: 10.26355/eurrev_202203_28369. Eur Rev Med Pharmacol Sci. 2022. PMID: 35363371 Free PMC article. Review.
-
Reduction of synaptojanin 1 accelerates Aβ clearance and attenuates cognitive deterioration in an Alzheimer mouse model.J Biol Chem. 2013 Nov 1;288(44):32050-63. doi: 10.1074/jbc.M113.504365. Epub 2013 Sep 19. J Biol Chem. 2013. PMID: 24052255 Free PMC article.
-
Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4 cells.Cell Death Dis. 2014 May 29;5(5):e1270. doi: 10.1038/cddis.2014.200. Cell Death Dis. 2014. PMID: 24874742 Free PMC article.
-
Autophagy and cancer cell metabolism.Semin Cell Dev Biol. 2012 Jun;23(4):395-401. doi: 10.1016/j.semcdb.2012.01.005. Epub 2012 Jan 18. Semin Cell Dev Biol. 2012. PMID: 22281437 Free PMC article. Review.
References
-
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
