

The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation
- PMID: 20202975
- PMCID: PMC2912657
- DOI: 10.1093/cvr/cvq076
The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation
Abstract
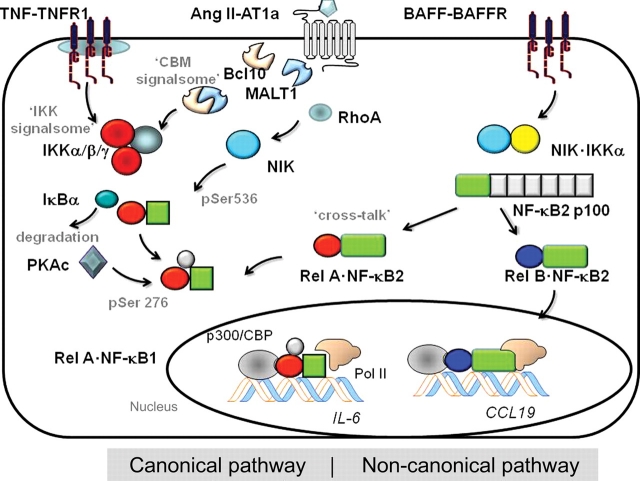
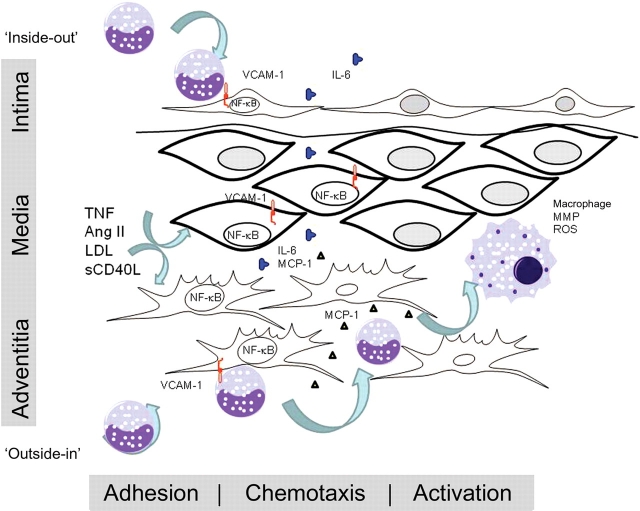
Vascular inflammation is a common pathophysiological response to diverse cardiovascular disease processes, including atherosclerosis, myocardial infarction, congestive heart failure, and aortic aneurysms/dissection. Inflammation is an ordered process initiated by vascular injury that produces enhanced leucocyte adherence, chemotaxis, and finally activation in situ. This process is coordinated by local secretion of adhesion molecules, chemotactic factors, and cytokines whose expression is the result of vascular injury-induced signal transduction networks. A wide variety of mediators of the vascular injury response have been identified; these factors include vasoactive peptides (angiotensin II, Ang II), CD40 ligands, oxidized cholesterol, and advanced glycation end-products. Downstream, the nuclear factor-kappaB (NF-kappaB) transcription factor performs an important signal integration step, responding to mediators of vascular injury in a stimulus-dependent and cell type-specific manner. The ultimate consequence of NF-kappaB signalling is the activation of inflammatory genes including adhesion molecules and chemotaxins. However, clinically, the hallmark of vascular NF-kappaB activation is the production of interleukin-6 (IL-6), whose local role in vascular inflammation is relatively unknown. The recent elucidation for the role of the IL-6 signalling pathway in Ang II-induced vascular inflammation as one that controls monocyte activation as well as its diverse signalling mechanism will be reviewed. These new discoveries further our understanding for the important role of the NF-kappaB-IL-6 signalling pathway in the process of vascular inflammation.
Figures
Similar articles
-
Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs.Physiol Genomics. 2002 Oct 2;11(1):21-30. doi: 10.1152/physiolgenomics.00062.2002. Physiol Genomics. 2002. PMID: 12361987
-
Angiotensin IV activates the nuclear transcription factor-kappaB and related proinflammatory genes in vascular smooth muscle cells.Circ Res. 2005 May 13;96(9):965-73. doi: 10.1161/01.RES.0000166326.91395.74. Epub 2005 Apr 14. Circ Res. 2005. PMID: 15831814
-
Quercetin disrupts tyrosine-phosphorylated phosphatidylinositol 3-kinase and myeloid differentiation factor-88 association, and inhibits MAPK/AP-1 and IKK/NF-κB-induced inflammatory mediators production in RAW 264.7 cells.Immunobiology. 2013 Dec;218(12):1452-67. doi: 10.1016/j.imbio.2013.04.019. Epub 2013 May 9. Immunobiology. 2013. PMID: 23735482
-
Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis.Adv Clin Chem. 2015;70:1-30. doi: 10.1016/bs.acc.2015.03.004. Epub 2015 Apr 22. Adv Clin Chem. 2015. PMID: 26231484 Review.
-
Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation.Rheumatology (Oxford). 2008 May;47(5):584-90. doi: 10.1093/rheumatology/kem298. Epub 2008 Jan 29. Rheumatology (Oxford). 2008. PMID: 18234712 Review.
Cited by
-
Rapid cyclic stretching induces a synthetic, proinflammatory phenotype in cultured human intestinal smooth muscle, with the potential to alter signaling to adjacent bowel cells.bioRxiv [Preprint]. 2024 Oct 15:2024.10.12.617767. doi: 10.1101/2024.10.12.617767. bioRxiv. 2024. PMID: 39464046 Free PMC article. Preprint.
-
Dopamine activates NF-κB and primes the NLRP3 inflammasome in primary human macrophages.Brain Behav Immun Health. 2020 Feb;2:100030. doi: 10.1016/j.bbih.2019.100030. Epub 2019 Dec 31. Brain Behav Immun Health. 2020. PMID: 33665636 Free PMC article.
-
Myeloid Differentiation Primary Response 88-Cyclin D1 Signaling in Breast Cancer Cells Regulates Toll-Like Receptor 3-Mediated Cell Proliferation.Front Oncol. 2020 Sep 18;10:1780. doi: 10.3389/fonc.2020.01780. eCollection 2020. Front Oncol. 2020. PMID: 33072559 Free PMC article.
-
Acetylated K676 TGFBIp as a severity diagnostic blood biomarker for SARS-CoV-2 pneumonia.Sci Adv. 2020 Jul 31;6(31):eabc1564. doi: 10.1126/sciadv.abc1564. Print 2020 Jul. Sci Adv. 2020. PMID: 32937590 Free PMC article.
-
Telmisartan Protects Against Aluminum-Induced Alzheimer-like Pathological Changes in Rats.Neurotox Res. 2020 Feb;37(2):275-285. doi: 10.1007/s12640-019-00085-z. Epub 2019 Jul 22. Neurotox Res. 2020. PMID: 31332715
References
-
- Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. doi:10.1161/01.ATV.0000214999.12921.4f. - DOI - PubMed
-
- Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. doi:10.1016/j.cardiores.2007.06.023. - DOI - PMC - PubMed
-
- Springer T. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multiple paradigm. Cell. 1994;76:301. doi:10.1016/0092-8674(94)90337-9. - DOI - PubMed
-
- Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi:10.1056/NEJMoa021993. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
