

Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling
- PMID: 20203212
- PMCID: PMC2864641
- DOI: 10.1523/JNEUROSCI.5712-09.2010
Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling
Abstract
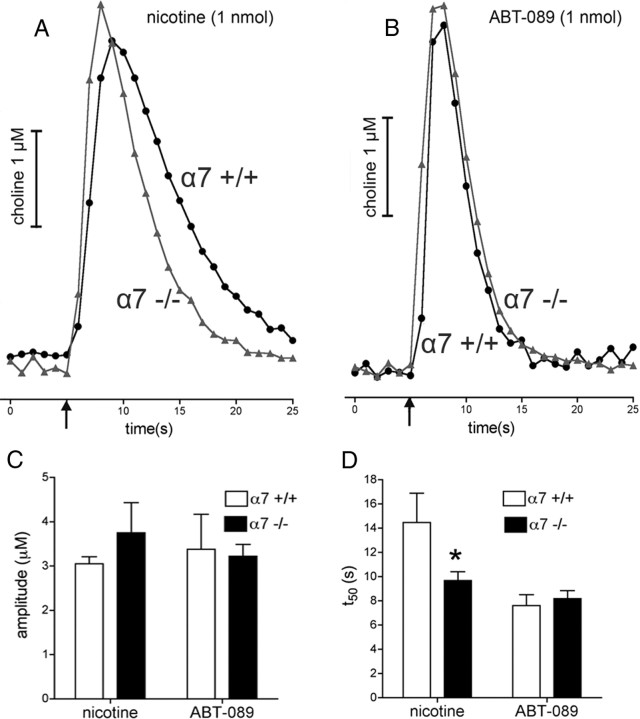
One-second-long increases in prefrontal cholinergic activity ("transients") were demonstrated previously to be necessary for the incorporation of cues into ongoing cognitive processes ("cue detection"). Nicotine and, more robustly, selective agonists at alpha4beta2* nicotinic acetylcholine receptors (nAChRs) enhance cue detection and attentional performance by augmenting prefrontal cholinergic activity. The present experiments determined the role of beta2-containing and alpha7 nAChRs in the generation of prefrontal cholinergic and glutamatergic transients in vivo. Transients were evoked by nicotine, the alpha4beta2* nAChR agonist ABT-089 [2-methyl-3-(2-(S)-pyrrolindinylmethoxy) pyridine dihydrochloride], or the alpha7 nAChR agonist A-582941 [2-methyl-5-(6-phenyl-pyridazin-3-yl)-octahydro-pyrrolo[3,4-c]pyrrole]. Transients were recorded in mice lacking beta2 or alpha7 nAChRs and in rats after removal of thalamic glutamatergic or midbrain dopaminergic inputs to prefrontal cortex. The main results indicate that stimulation of alpha4beta2* nAChRs evokes glutamate release and that the presence of thalamic afferents is necessary for the generation of cholinergic transients. ABT-089-evoked transients were completely abolished in mice lacking beta2* nAChRs. The amplitude, but not the decay rate, of nicotine-evoked transients was reduced by beta2* knock-out. Conversely, in mice lacking the alpha7 nAChR, the decay rate, but not the amplitude, of nicotine-evoked cholinergic and glutamatergic transients was attenuated. Substantiating the role of alpha7 nAChR in controlling the duration of release events, stimulation of alpha7 nAChR produced cholinergic transients that lasted 10- to 15-fold longer than those evoked by nicotine. alpha7 nAChR-evoked cholinergic transients are mediated in part by dopaminergic activity. Prefrontal alpha4beta2* nAChRs play a key role in evoking and facilitating the transient glutamatergic-cholinergic interactions that are necessary for cue detection and attentional performance.
Figures







Similar articles
-
Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex.J Neurosci. 2008 Apr 2;28(14):3769-80. doi: 10.1523/JNEUROSCI.5251-07.2008. J Neurosci. 2008. PMID: 18385335 Free PMC article.
-
Enhancement of attentional performance by selective stimulation of alpha4beta2(*) nAChRs: underlying cholinergic mechanisms.Neuropsychopharmacology. 2010 May;35(6):1391-401. doi: 10.1038/npp.2010.9. Epub 2010 Feb 10. Neuropsychopharmacology. 2010. PMID: 20147893 Free PMC article.
-
Selective potentiation of (α4)3(β2)2 nicotinic acetylcholine receptors augments amplitudes of prefrontal acetylcholine- and nicotine-evoked glutamatergic transients in rats.Biochem Pharmacol. 2013 Nov 15;86(10):1487-96. doi: 10.1016/j.bcp.2013.09.005. Epub 2013 Sep 16. Biochem Pharmacol. 2013. PMID: 24051136 Free PMC article.
-
The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus.Biochem Pharmacol. 2015 Oct 15;97(4):439-444. doi: 10.1016/j.bcp.2015.07.015. Epub 2015 Jul 23. Biochem Pharmacol. 2015. PMID: 26212541 Free PMC article. Review.
-
nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms.Biochem Pharmacol. 2009 Oct 1;78(7):658-67. doi: 10.1016/j.bcp.2009.04.019. Epub 2009 May 4. Biochem Pharmacol. 2009. PMID: 19406107 Free PMC article. Review.
Cited by
-
Dystonia and levodopa-induced dyskinesias in Parkinson's disease: Is there a connection?Neurobiol Dis. 2019 Dec;132:104579. doi: 10.1016/j.nbd.2019.104579. Epub 2019 Aug 22. Neurobiol Dis. 2019. PMID: 31445160 Free PMC article. Review.
-
Chemogenetic inhibition of prefrontal projection neurons constrains top-down control of attention in young but not aged rats.Brain Struct Funct. 2021 Sep;226(7):2357-2373. doi: 10.1007/s00429-021-02336-2. Epub 2021 Jul 10. Brain Struct Funct. 2021. PMID: 34247267 Free PMC article.
-
Amperometric measurement of glutamate release modulation by gabapentin and pregabalin in rat neocortical slices: role of voltage-sensitive Ca2+ α2δ-1 subunit.J Pharmacol Exp Ther. 2011 Jul;338(1):240-5. doi: 10.1124/jpet.110.178384. Epub 2011 Apr 4. J Pharmacol Exp Ther. 2011. PMID: 21464332 Free PMC article.
-
Nicotinic Acetylcholine Receptor α7 Subunit Is an Essential Regulator of Seizure Susceptibility.Front Neurol. 2021 Apr 12;12:656752. doi: 10.3389/fneur.2021.656752. eCollection 2021. Front Neurol. 2021. PMID: 33912128 Free PMC article.
-
Paternal nicotine enhances fear memory, reduces nicotine administration, and alters hippocampal genetic and neural function in offspring.Addict Biol. 2021 Jan;26(1):e12859. doi: 10.1111/adb.12859. Epub 2019 Nov 28. Addict Biol. 2021. PMID: 31782218 Free PMC article.
References
-
- Arnold HM, Burk JA, Hodgson EM, Sarter M, Bruno JP. Differential cortical acetylcholine release in rats performing a sustained attention task versus behavioral control tasks that do not explicitly tax attention. Neuroscience. 2002;114:451–460. - PubMed
-
- Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, Decker MW, Frost JM, Gronlien JH, Gubbins E, Li J, Malysz J, Markosyan S, Marsh K, Meyer MD, Nikkel AL, Radek RJ, Robb HM, Timmermann D, Sullivan JP, Gopalakrishnan M. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci. 2007;27:10578–10587. - PMC - PubMed
-
- Buccafusco JJ, Jackson WJ, Terry AV, Jr, Marsh KC, Decker MW, Arneric SP. Improvement in performance of a delayed matching-to-sample task by monkeys following ABT-418: a novel cholinergic channel activator for memory enhancement. Psychopharmacology (Berl) 1995;120:256–266. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources