

Mutations in FLVCR2 are associated with proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (Fowler syndrome)
- PMID: 20206334
- PMCID: PMC2833392
- DOI: 10.1016/j.ajhg.2010.02.004
Mutations in FLVCR2 are associated with proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (Fowler syndrome)
Abstract
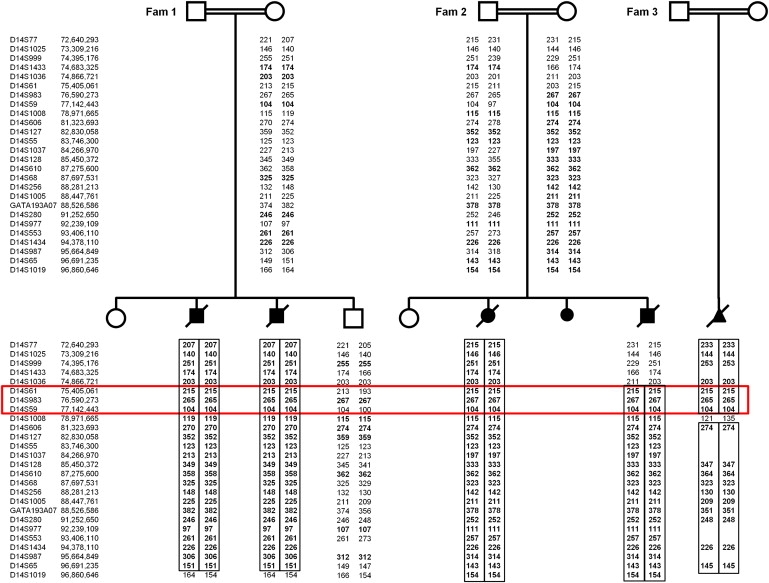
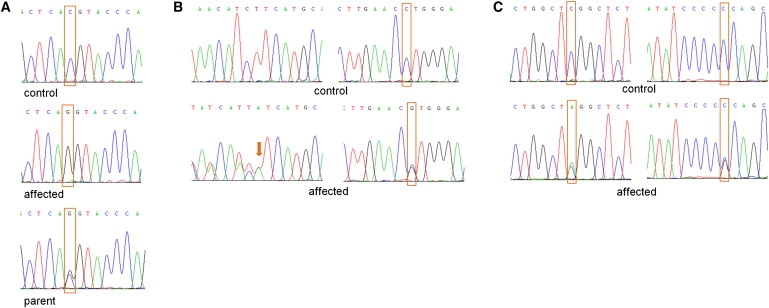
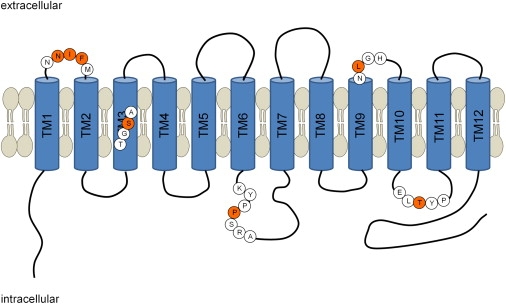
Proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (PVHH), also known as Fowler syndrome, is an autosomal-recessively inherited prenatal lethal disorder characterized by hydranencephaly; brain stem, basal ganglia, and spinal cord diffuse clastic ischemic lesions with calcifications; glomeruloid vasculopathy of the central nervous system and retinal vessels; and a fetal akinesia deformation sequence (FADS) with muscular neurogenic atrophy. To identify the molecular basis for Fowler syndrome, we performed autozygosity mapping studies in three consanguineous families. The results of SNP microarrays and microsatellite marker genotyping demonstrated linkage to chromosome 14q24.3. Direct sequencing of candidate genes within the target interval revealed five different germline mutations in FLVCR2 in five families with Fowler syndrome. FLVCR2 encodes a transmembrane transporter of the major facilitator superfamily (MFS) hypothesized to be involved in regulation of growth, calcium exchange, and homeostasis. This is the first gene to be associated with Fowler syndrome, and this finding provides a basis for further studies to elucidate the pathogenetic mechanisms and phenotypic spectrum of associated disorders.
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Fowler M., Dow R., White T.A., Greer C.H. Congenital hydrocephalus-hydrencephaly in five siblings, with autopsy studies: a new disease. Dev. Med. Child Neurol. 1972;14:173–188. - PubMed
-
- Harper C., Hockey A. Proliferative vasculopathy and an hydranencephalic-hydrocephalic syndrome: a neuropathological study of two siblings. Dev. Med. Child Neurol. 1983;25:232–239. - PubMed
-
- Norman M.G., McGillivray B. Fetal neuropathology of proliferative vasculopathy and hydranencephaly-hydrocephaly with multiple limb pterygia. Pediatr. Neurosci. 1988;14:301–306. - PubMed
-
- Harding B.N., Ramani P., Thurley P. The familial syndrome of proliferative vasculopathy and hydranencephaly-hydrocephaly: immunocytochemical and ultrastructural evidence for endothelial proliferation. Neuropathol. Appl. Neurobiol. 1995;21:61–67. - PubMed
-
- Castro-Gago M., Pintos-Martínez E., Forteza-Vila J., Iglesias-Diz M., Ucieda-Somoza R., Silva-Villar I., Codesido-López J., Viso-Lorenzo A., Campos Y., Arenas J., Eirís-Puñal J. Congenital hydranencephalic-hydrocephalic syndrome with proliferative vasculopathy: a possible relation with mitochondrial dysfunction. J. Child Neurol. 2001;16:858–862. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
