

Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation
- PMID: 20207196
- PMCID: PMC2976657
- DOI: 10.1016/j.molmed.2010.02.002
Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation
Abstract
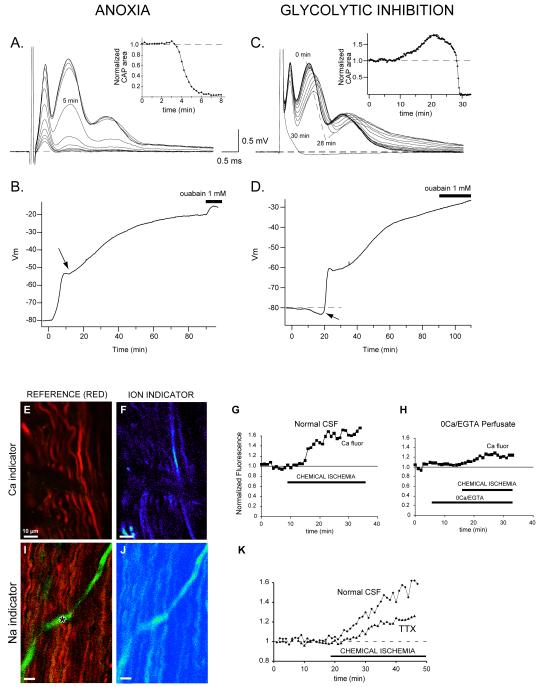
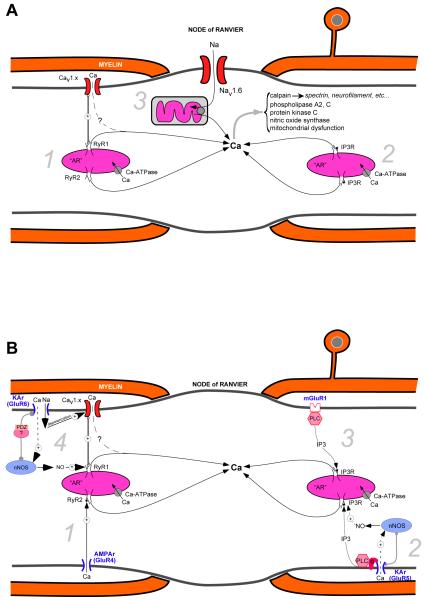
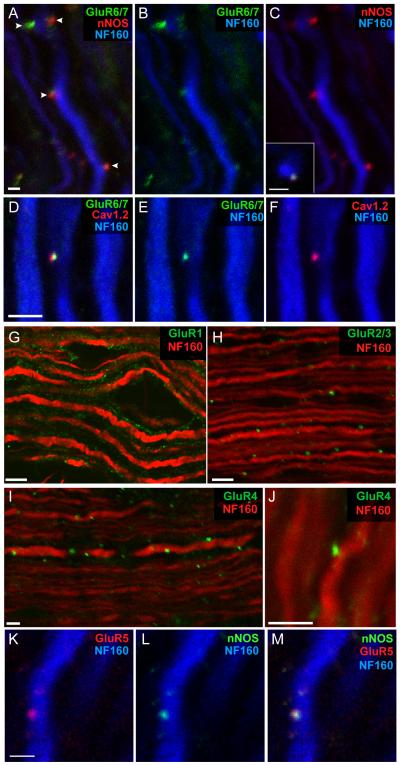
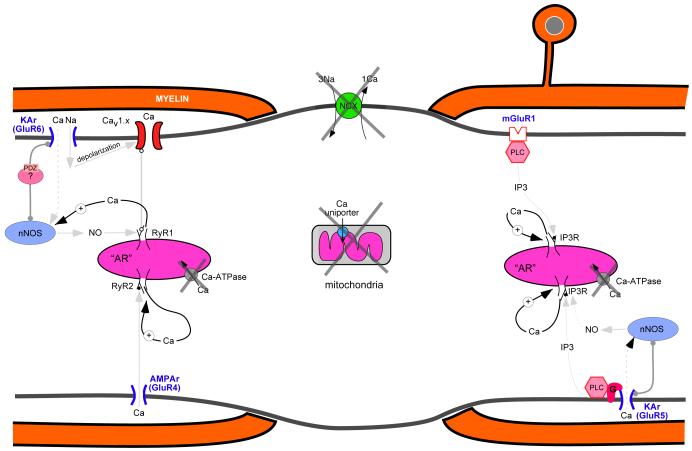
Axonal degeneration causes morbidity in many neurological conditions including stroke, neurotrauma and multiple sclerosis. The limited ability of central nervous system (CNS) neurons to regenerate, combined with the observation that axonal damage causes clinical disability, has spurred efforts to investigate the mechanisms of axonal degeneration. Ca influx from outside the axon is a key mediator of injury. More recently, substantial pools of intra-axonal Ca sequestered in the 'axoplasmic reticulum' have been reported. These Ca stores are under the control of multimolecular 'nanocomplexes' located along the internodes under the myelin. The overactivation of these complexes during disease can lead to a lethal release of Ca from intra-axonal stores. Rich receptor pharmacology offers tantalizing therapeutic options targeting these nanocomplexes in the many diseases where axonal degeneration is prominent.
Figures
References
-
- Waxman SG, Kocsis JD, Stys PK. The Axon : structure, function and pathophysiology. Oxford University Press; New York; Oxford: 1995. p. xv.p. 692. [2] of col. plates.
-
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8(3):280–91. - PubMed
-
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233(1-2):3–13. - PubMed
-
- Wilkins A, Scolding N. Protecting axons in multiple sclerosis. Mult Scler. 2008;14(8):1013–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
