

Nucleosome dynamics define transcriptional enhancers
- PMID: 20208536
- PMCID: PMC2932437
- DOI: 10.1038/ng.545
Nucleosome dynamics define transcriptional enhancers
Abstract
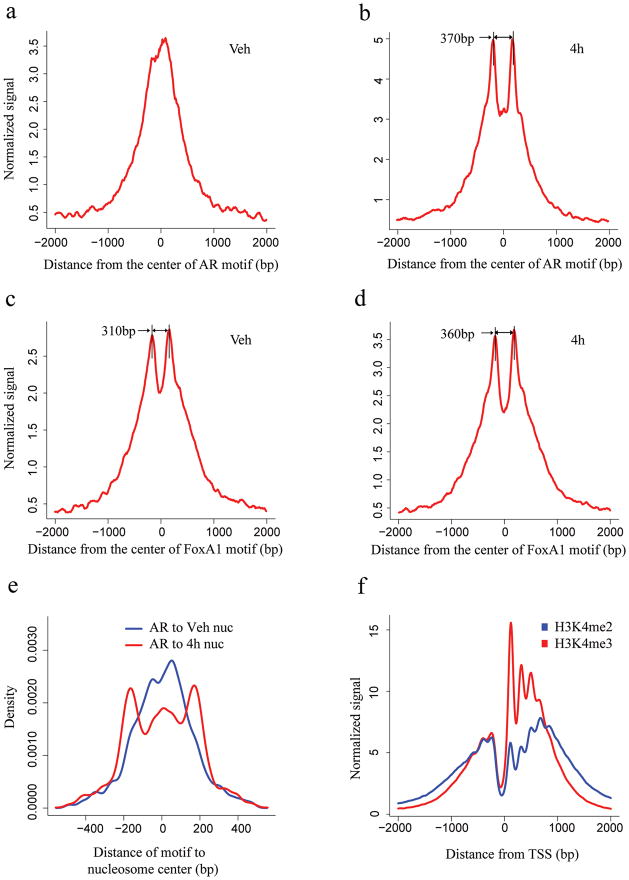
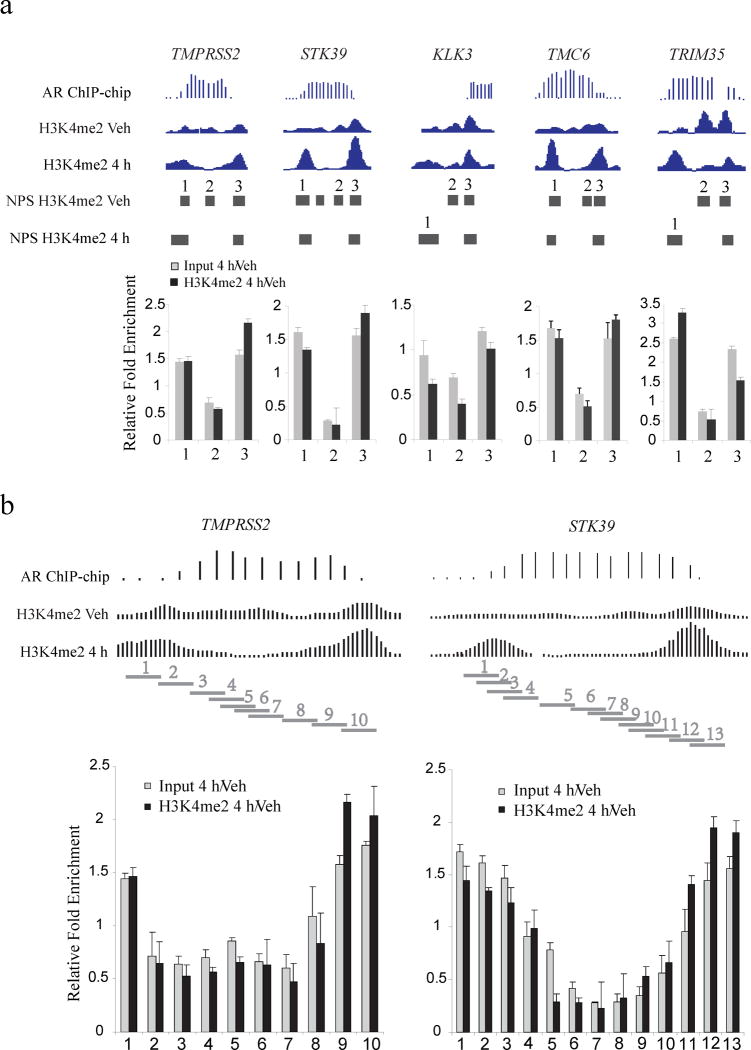
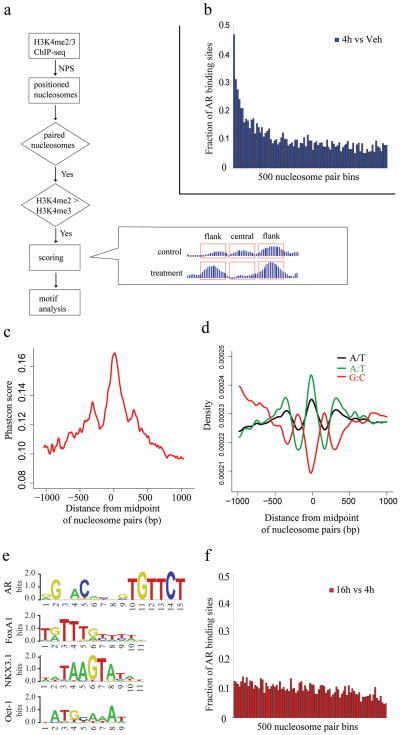
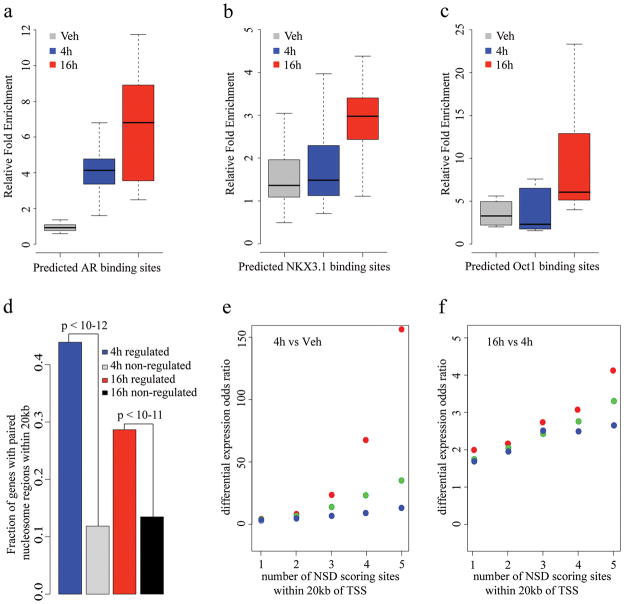
Chromatin plays a central role in eukaryotic gene regulation. We performed genome-wide mapping of epigenetically marked nucleosomes to determine their position both near transcription start sites and at distal regulatory elements, including enhancers. In prostate cancer cells, where androgen receptor binds primarily to enhancers, we found that androgen treatment dismisses a central nucleosome present at androgen receptor binding sites that is flanked by a pair of marked nucleosomes. A new quantitative model built on the behavior of such nucleosome pairs correctly identified regions bound by the regulators of the immediate androgen response, including androgen receptor and FOXA1. More importantly, this model also correctly predicted previously unidentified binding sites for other transcription factors present after prolonged androgen stimulation, including OCT1 and NKX3-1. Therefore, quantitative modeling of enhancer structure provides a powerful predictive method to infer the identity of transcription factors involved in cellular responses to specific stimuli.
Figures
Comment in
-
Epigenetic marks identify functional elements.Nat Genet. 2010 Apr;42(4):282-4. doi: 10.1038/ng0410-282. Nat Genet. 2010. PMID: 20348962
References
-
- Yuan GC, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
