

Calcium signaling and amyloid toxicity in Alzheimer disease
- PMID: 20212036
- PMCID: PMC2857063
- DOI: 10.1074/jbc.R109.080895
Calcium signaling and amyloid toxicity in Alzheimer disease
Abstract
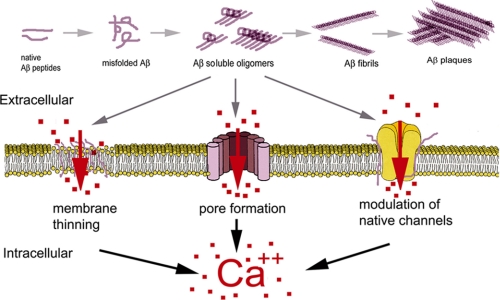
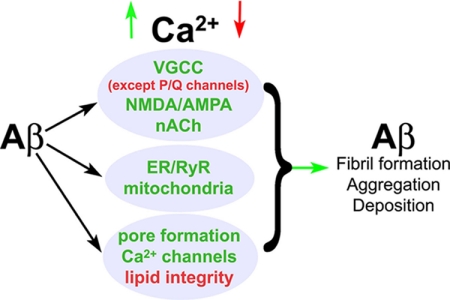
Intracellular Ca(2+) signaling is fundamental to neuronal physiology and viability. Because of its ubiquitous roles, disruptions in Ca(2+) homeostasis are implicated in diverse disease processes and have become a major focus of study in multifactorial neurodegenerative diseases such as Alzheimer disease (AD). A hallmark of AD is the excessive production of beta-amyloid (Abeta) and its massive accumulation in amyloid plaques. In this minireview, we highlight the pathogenic interactions between altered cellular Ca(2+) signaling and Abeta in its different aggregation states and how these elements coalesce to alter the course of the neurodegenerative disease. Ca(2+) and Abeta intersect at several functional levels and temporal stages of AD, thereby altering neurotransmitter receptor properties, disrupting membrane integrity, and initiating apoptotic signaling cascades. Notably, there are reciprocal interactions between Ca(2+) pathways and amyloid pathology; altered Ca(2+) signaling accelerates Abeta formation, whereas Abeta peptides, particularly in soluble oligomeric forms, induce Ca(2+) disruptions. A degenerative feed-forward cycle of toxic Abeta generation and Ca(2+) perturbations results, which in turn can spin off to accelerate more global neuropathological cascades, ultimately leading to synaptic breakdown, cell death, and devastating memory loss. Although no cause or cure is currently known, targeting Ca(2+) dyshomeostasis as an underlying and integral component of AD pathology may result in novel and effective treatments for AD.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
